Los trastornos hematológicos se describen como una complicación rara de la infección aguda por el virus de Epstein-Barr (VEB). La pancitopenia, en un paciente con una infección por VEB, debe obligar a descartar una inmunodeficiencia, un síndrome hemofagocítico (SH) o un proceso linfoproliferativo, como en los 2 casos presentados en este artículo1,2.
Caso 1El primero corresponde a una niña de 12 años, que presentaba tos y afonía de 2 meses de evolución, con sensación distérmica intermitente y epistaxis ocasional, se detectó tumor en cavum. Ingresa en la unidad de cuidados intensivos pediátricos (UCIP), tras exéresis de dicho proceso tumoral. En la exploración destaca esplenomegalia masiva, con hepatomegalia de 2 traveses y adenopatías laterocervicales.
Exploraciones complementarias normales, excepto pancitopenia y reacción en cadena de la polimerasa (PCR) a VEB positiva (15.000 copias/ml). La tomografía computarizada (TC) toracoabdominal muestra adenopatías laterocervicales, hiliares y mesentéricas con hepatomegalia de 5cm y una esplenomegalia de 26cm.
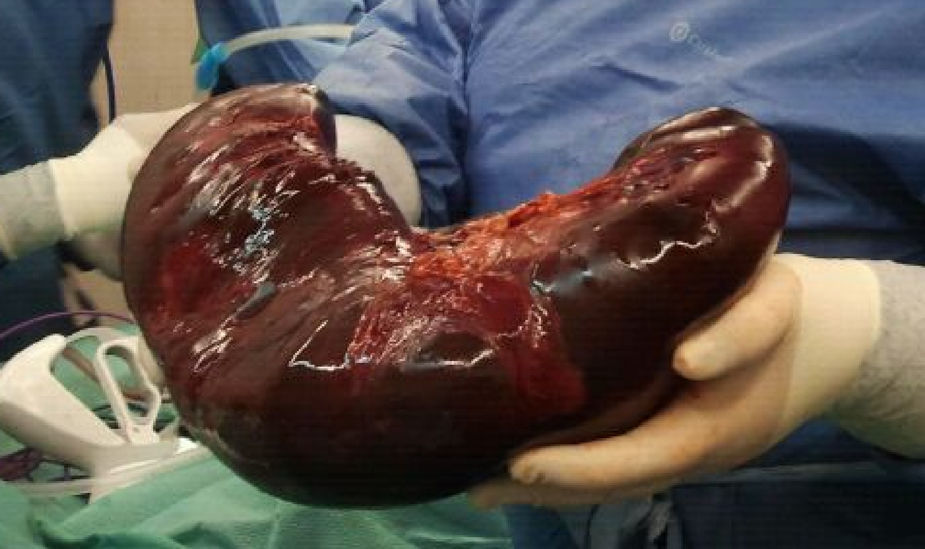
A su ingreso en la UCIP presenta shock hemorrágico secundario a sangrado de cavum. Desarrolla un cuadro de distrés respiratorio agudo (SDRA), que requiere ventilación mecánica de alta frecuencia, así como una insuficiencia renal aguda, que precisa de depuración extrarrenal. La esplenomegalia aumenta de tamaño progresivamente, y origina un síndrome compartimental intraabdominal. Se procede a embolización esplénica y posterior esplenectomía (fig. 1), con recuperación progresiva del fallo multiorgánico. Al tercer día, inicia cuadro de fiebre persistente, con una elevación de la carga viral en plasma de VEB hasta 750.000copias/ml, ferritina de hasta 46.000μg/l, fibrinógeno mínimo de 1,1g/l, LDH máxima de 6.600UI/l y triglicéridos máximos de 1.100mg/dl, con actividad citotóxica natural killer (NK) disminuida, compatibles con SH. Simultáneamente, las biopsias realizadas de cavum y de una adenopatía laterocervical, permiten el diagnóstico de síndrome linfoproliferativo T asociado a VEB de la infancia. El estudio de extensión demuestra infiltración medular por linfocitos T CD8. Se inicia tratamiento quimioterápico con ganciclovir (12mg/kg/día), rituximab (375mg/m2/dosis) y gammaglobulinas inespecíficas (0,5g/kg/dosis) para el VEB, así como tratamiento del SH según protocolo Hemophagocytic Lymphohistiocytosis (HLH)-2004 con dexametasona (10mg/m2/día), etopósido (100mg/m2/dosis) y ciclosporina A (6mg/kg/día). Tras la falta de remisión, se inicia infusión de globulina antitimocítica (5mg/kg/día) y, posteriormente, 5 sesiones de plasmaféresis, hasta conseguir buena respuesta clínico-analítica.
 tras esplenectomía del caso 1.")
Se completa el estudio de formas de inmunodeficiencia asociadas a VEB-HLH y linfoproliferación (fenotipado linfocitario T, población NKT, expresión CD27, inmunoglobulinas, estudio degranulación y expresión de perforinas, estudio gen ITK, que resultan normales; solo se puede objetivar la citotoxicidad NK disminuida en fase aguda del SH). Tras 3 meses de iniciado el cuadro, y dada la no resolución del mismo, a pesar del correcto tratamiento, se procede al trasplante de progenitores hematopoyéticos (TPH) de donante emparentado, que transcurre sin incidencias, con normalización clínica posterior.
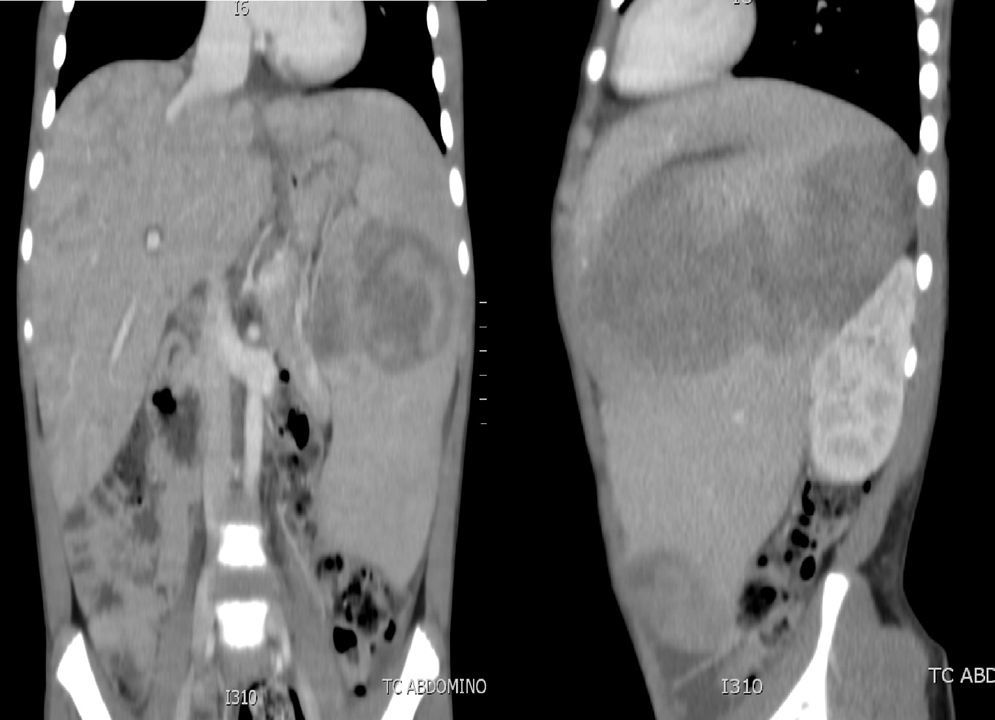
Caso 2El segundo caso corresponde a un niño de 10 años, que consulta a urgencias por un cuadro de un mes de evolución de fiebre, somnolencia, distensión abdominal y pérdida de peso no cuantificada. En la exploración física destaca importante hepatoesplenomegalia, y en la analítica se detecta pancitopenia, hiponatremia y disfunción hepática. Antígeno paludismo negativo y punción lumbar normal. Se solicita TC abdominal, donde se observa hepatoesplenomegalia de gran tamaño, con nódulos en su interior (fig. 2) y TC cerebral normal. Se realiza aspirado de médula ósea en el que no se observan blastos, pero sí imágenes de hemofagocitosis. Analítica sanguínea con ferritina elevada (33.000μg/l), LDH de 10.300UI/l, triglicéridos de 400mg/dl, fibrinógeno de 1,2g/l y carga viral para VEB superior a 2.600.000 copias/ml, por lo que se orienta como SH secundario a infección por VEB. Se inicia tratamiento con ganciclovir intravenoso (12mg/kg/día), gammaglobulinas inespecíficas (0,5g/kg/dosis) y tratamiento para el SH con etopósido (100mg/m2/dosis), ciclosporina (6mg/kg/día) y dexametasona (10mg/m2/día), previa realización de biopsia hepática. La muestra es diagnóstica de síndrome linfoproliferativo T/NK. Se completa el estudio de formas de inmunodeficiencia asociadas a VEB-HLH en el varón que, además de las descritas anteriormente para el caso 1, incluyen el estudio genético de los genes SH2D1A y BIRC4, que resultaron normales. El paciente presenta una evolución clínica y analítica inicial satisfactoria. A los 20 días se detecta una reactivación del SH, hepatoesplenomegalia masiva, reaparición de fiebre y elevación del número de copias de VEB. Se intensifica el tratamiento con rituximab (350mg/m2/dosis), plasmaféresis, foscarnet (180mg/kg/día) y tratamiento quimioterápico para el síndrome linfoproliferativo. El paciente presenta una hemorragia pulmonar grave que requiere intubación, fármacos vasoactivos, y que se atribuye a la activación del SH. Los parámetros de hemofagocitosis y la carga viral del VEB siguieron aumentando (ferritina máxima 707.300μg/l, LDH 12.000UI/l, triglicéridos 426mg/dl, carga viral máxima de 5.000.000 copias/ml), a pesar de la gammaglobulina antitimocítica (5mg/kg/dosis) y una dosis de tocilizumab (210mg/m2/dosis). Evoluciona hacia un SDRA refractario a todas las estrategias de ventilación, siendo exitus 2 meses después de su ingreso en la UCIP.

El SH se caracteriza por una proliferación histiocítica benigna, generalizada con hemofagocitosis en bazo, médula ósea, ganglios linfáticos e hígado. Se cree que es el resultado de un desequilibrio inmunológico generado por una hiperreactividad de los linfocitos T, que condiciona un estado de hipercitoquinemia que lleva a una activación exagerada de los macrófagos. Para su diagnóstico se exige el cumplimiento de una serie de criterios3, y es muy útil la determinación de la actividad citotóxica de las células NK4. El SH puede ser primario (o de base genética por defectos en genes relacionados con la función citotóxica de las células NK), o asociado a algún factor desencadenante, como las infecciones5. El VEB ha sido uno de los más vinculados al SH, tanto primario como secundario, tal y como se describe en nuestros casos y otros publicados en la literatura1,6,7. El tratamiento se basa en 2 pilares: el control de la causa subyacente (en nuestros casos la infección por VEB, mediante la combinación de antivirales y rituximab) y el control del estado hiperinflamatorio, seguido de la corrección del defecto genético mediante el TPH en los casos primarios. La fase inicial del tratamiento busca frenar la proliferación y activación de células T, mediante la combinación de dexametasona, ciclosporina y etopósido durante 8 semanas, según el protocolo HLH-2004. Otros tratamientos de segunda línea incluyen la globulina anti-timocítica y el anticuerpo monoclonal anti-CD523,8–10. En los casos presentados en este artículo se utilizaron varios escalones terapéuticos, aunque con diferente respuesta.
Es necesario un diagnóstico precoz para tratar rápida y enérgicamente la infección por VEB, y controlar el SH, de cara a aumentar la supervivencia de estos pacientes.
FinanciaciónLos autores declaran que no han recibido ninguna financiación.
Conflicto de interesesLos autores declaran que no tienen conflicto de intereses.