





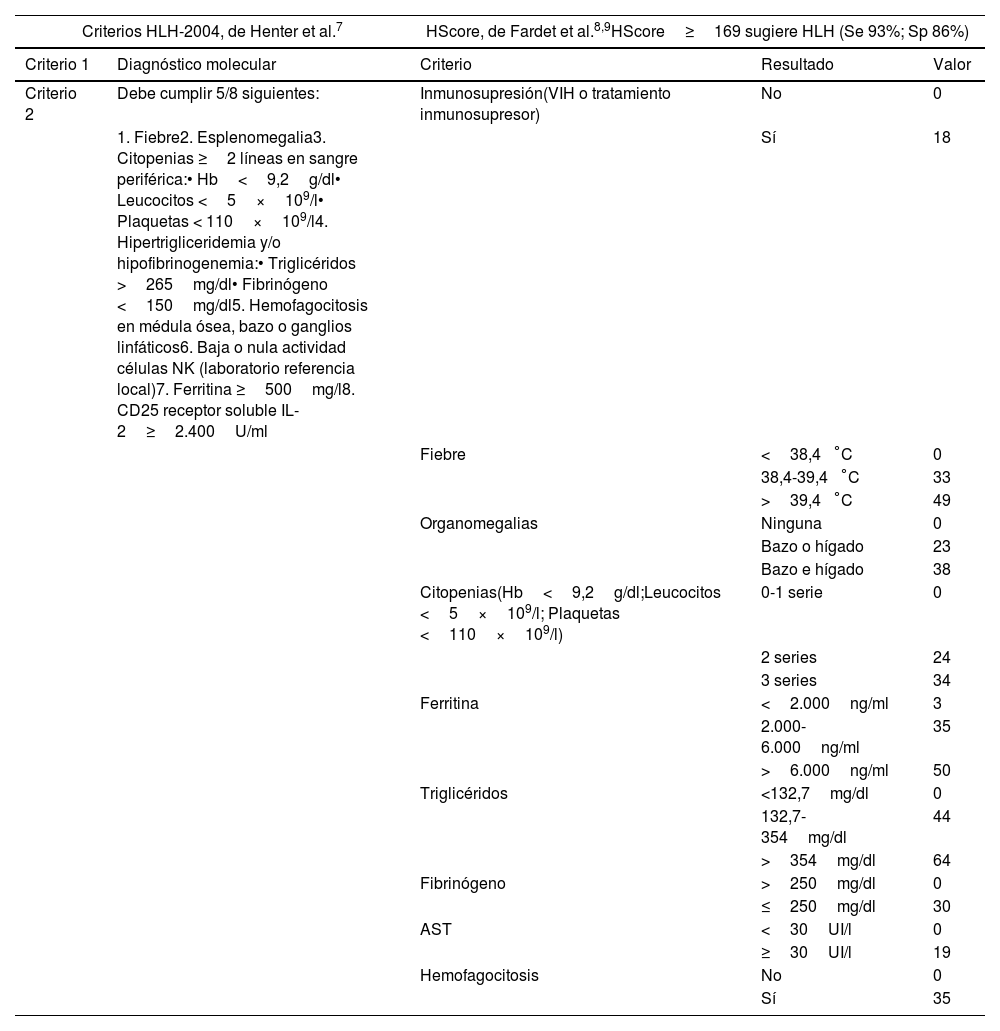

En los últimos años se ha producido un incremento de pacientes afectos por enfermedades oncohematológicas en los países desarrollados, secundario a la mejora en las tasas de supervivencia y la calidad de vida. Este aumento ha generado una mayor necesidad de asistencia de los mismos en los servicios de medicina intensiva (SMI), principalmente debido a complicaciones relacionadas con inmunosupresión, la toxicidad del tratamiento o complicaciones derivadas del propio cáncer. La inmunoterapia ha transformado el tratamiento del cáncer, pero puede causar efectos secundarios graves como el síndrome de liberación de citocinas y el síndrome hemofagocítico, los cuales a menudo requieren ingreso en los SMI. Esta revisión busca expandir el conocimiento y las estrategias de manejo de estas complicaciones en los SMI.
In recent years, there has been an increase in the number of patients affected by oncohematologic diseases in developed countries due to the improved survival rates and quality of life. This increase has generated a greater need for care in intensive care units (ICU), mainly due to complications related to immunosuppression, treatment toxicity or complications derived from cancer itself. Immunotherapy has transformed cancer treatment, but it can cause serious side effects such as cytokine release syndrome and hemophagocytic syndrome, which often require ICU admission. This review seeks to expand knowledge and management strategies for these complications in the ICU.
Artículo
Diríjase al área de socios de la web de la SEMICYUC (www.semicyuc.org ) y pulse el enlace a la revista.


