









Paroxysmal sympathetic hyperactivity (PSH) is a potentially life-threatening neurological emergency secondary to multiple acute acquired brain injuries. It is clinically characterized by the cyclic and simultaneous appearance of signs and symptoms secondary to exacerbated sympathetic discharge. The diagnosis is based on the clinical findings, and high alert rates are required. No widely available and validated homogeneous diagnostic criteria have been established to date. There have been recent consensus attempts to shed light on this obscure phenomenon. Its physiopathology is complex and has not been fully clarified. However, the excitation-inhibition model is the theory that best explains the different aspects of this condition, including the response to treatment with the available drugs. The key therapeutic references are the early recognition of the disorder, avoiding secondary injuries and the triggering of paroxysms. Once sympathetic crises occur, they must be peremptorily aborted and prevented. The later the syndrome is recognized, the poorer the patient outcome.
La hiperactividad simpática paroxística es una urgencia neurológica potencialmente letal secundaria a múltiples lesiones cerebrales agudas adquiridas. Se caracteriza por rasgos clínicos de aparición cíclica y simultánea, consecuencia de una descarga simpática exacerbada. El diagnóstico es clínico, requiriendo elevados índices de alerta. Actualmente no existen criterios diagnósticos homogéneos que estén ampliamente difundidos y validados. El consenso reciente intenta arrojar luz sobre este oscuro panorama. Su fisiopatología es compleja y aún no ha sido elucidada con certeza; sin embargo, la teoría basada en el modelo excitación-inhibición es la que mejor explica los distintos aspectos de esta entidad, incluyendo la respuesta a la terapia con los fármacos disponibles. Los pilares terapéuticos se asientan sobre el reconocimiento precoz, evitar insultos secundarios y el desencadenamiento de los paroxismos. De ocurrir crisis simpáticas, es que estas se aborten de forma perentoria y que se prevengan. Cuanto más tarde en reconocerse el síndrome, peores serán los resultados.
In 1929 Wilder Penfield described the case of a 41-year-old woman who developed sudden and paroxysmal arterial hypertension, tachycardia and tachypnea with occasional changes from normal ventilation toward Cheyne-Stokes respiration.1 During the paroxysmal episodes the body temperature was seen to rise, with intermittent dilatation and contraction of the pupils, and excessive tearing.1 The patient presented a third ventricle cholesteatoma. Penfield referred to these episodes as “diencephalic autonomic epilepsy”, and suggested that they reflected hyperactivity of both the sympathetic and the parasympathetic autonomic nervous system.1
Since this first publication there have been reports of similar conditions in many types of serious brain injuries, and different terms have been used to describe them.2–6 Sympathetic alterations predominate in some cases and parasympathetic alterations in others, though both the sympathetic and the parasympathetic autonomic nervous system can be more or less equally affected in some patients. This circumstance tends to cause confusion, making it difficult to adequately study the disease2,6 (Table 1).
Different forms of paroxysmal sympathetic hyperactivity (PSH) nomenclature over time.
| Diencephalic seizures |
| Brainstem attack |
| Central autonomic dysregulation |
| Hyperadrenergic state |
| Midbrain syndrome |
| Decerebration tonic spasms |
| Cerebellar tonic discharges |
| Sympathetic adrenal response |
| Autonomic dysfunction syndrome |
| Hypothalamic-midbrain dysregulation syndrome |
| Dysautonomy |
| Hyperpyrexia with prolonged muscle contraction |
| Autonomic storm |
| Sympathetic storm |
| Paroxysmal autonomic instability with dystonia |
| Paroxysmal autonomic dysregulation |
| Paroxysmal sympathetic hyperactivity |
The term “paroxysmal sympathetic hyperactivity” (PSH) has recently been introduced, summarizing the main characteristics of the syndrome, which results from overactivity of only the sympathetic nervous system.2–5,7,8
Paroxysmal sympathetic hyperactivity is a genuine neurological emergency that may go undetected if not taken into account.2–8 The diagnosis is mainly established on an exclusion basis, ruling out other possible diagnoses, and requires a strong degree of suspicion. Failure to adequately detect and treat the condition is associated to high mortality-morbidity rates.2–5
The main objective of this study is to offer simple and practical answers to questions referred to the correct identification and management of the disease.
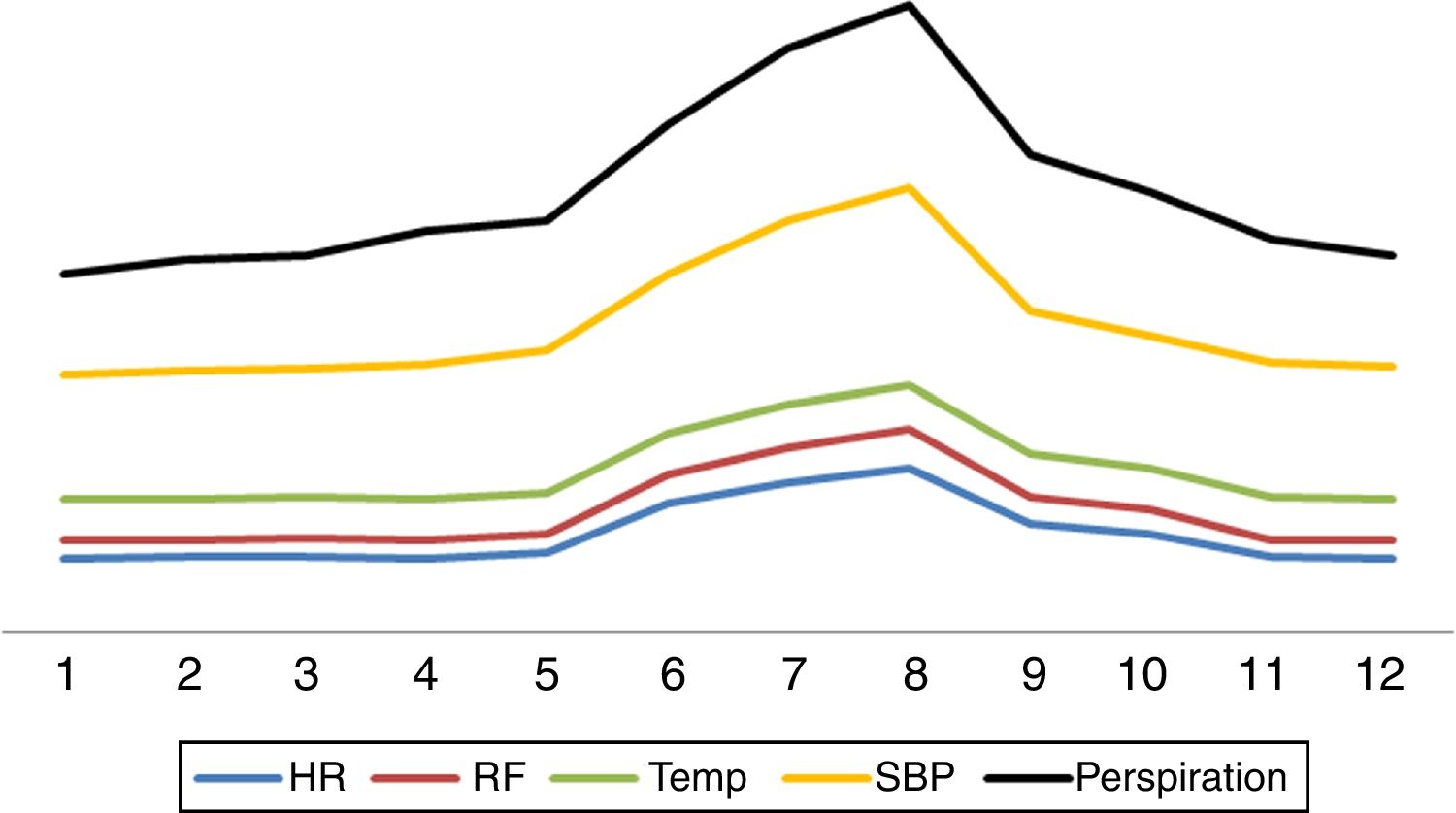
Question 1. What do we mean by paroxysmal sympathetic hyperactivity?Paroxysmal sympathetic hyperactivity comprises a series of signs and symptoms reflecting exacerbated sympathetic activity, including: tachycardia, arterial hypertension, tachypnea, hyperthermia, generalized perspiration, anomalous motor activity (dystonia, muscle stiffness, extension) and mechanical ventilator maladjustment.2–8 These clinical features moreover manifest simultaneously.2–8 The number of signs or symptoms is closely correlated to the ultimate outcome,4 with more profuse manifestations being associated to a poorer prognosis.4
Paroxysmal sympathetic hyperactivity is not a primary disorder but always develops as a result of brain injuries of variable nature and severity.2–8
Question 2. What are the main characteristics of paroxysmal sympathetic hyperactivity?Paroxysmal sympathetic hyperactivity manifests abruptly in cyclic episodes, either spontaneously or in response to stimuli such as pain, bathing, the aspiration of secretions, exposure to light, touch or physiotherapy.2–8 Sympathetic hyperactivity can manifest at any time in the course of the disorder that causes it, though it is usually detected after the first week, coinciding with a decrease or the suspension of deep sedoanalgesia2–8 (Fig. 1). The literature indicates that in most instances the diagnosis is made one week after patient admission.8 The paroxysms appear 3–5 times a day and last an average of 30min.2–8
The syndrome has three evolutive stages.8 Hyperacute phase I covers the first week, during which the expression of brain damage is maximum. The patient is generally unstable and intensive treatment is provided, including generally deep sedation and analgesia. As a result, the diagnosis cannot be established in this stage, unless a waking test is made for some reason or the patient accidentally emerges from anesthesia.8 In phase II the syndrome – with the aforementioned features – is fully expressed. This stage typically extends to two and a half months after the causal injury (day 74).
Cessation of the perspiration episodes marks the end of stage II,8 which is followed by stage III. The latter comprises the rehabilitation period and can last for years – though the episodes in this case are generally less frequent and of lesser intensity and duration8 (Fig. 2).
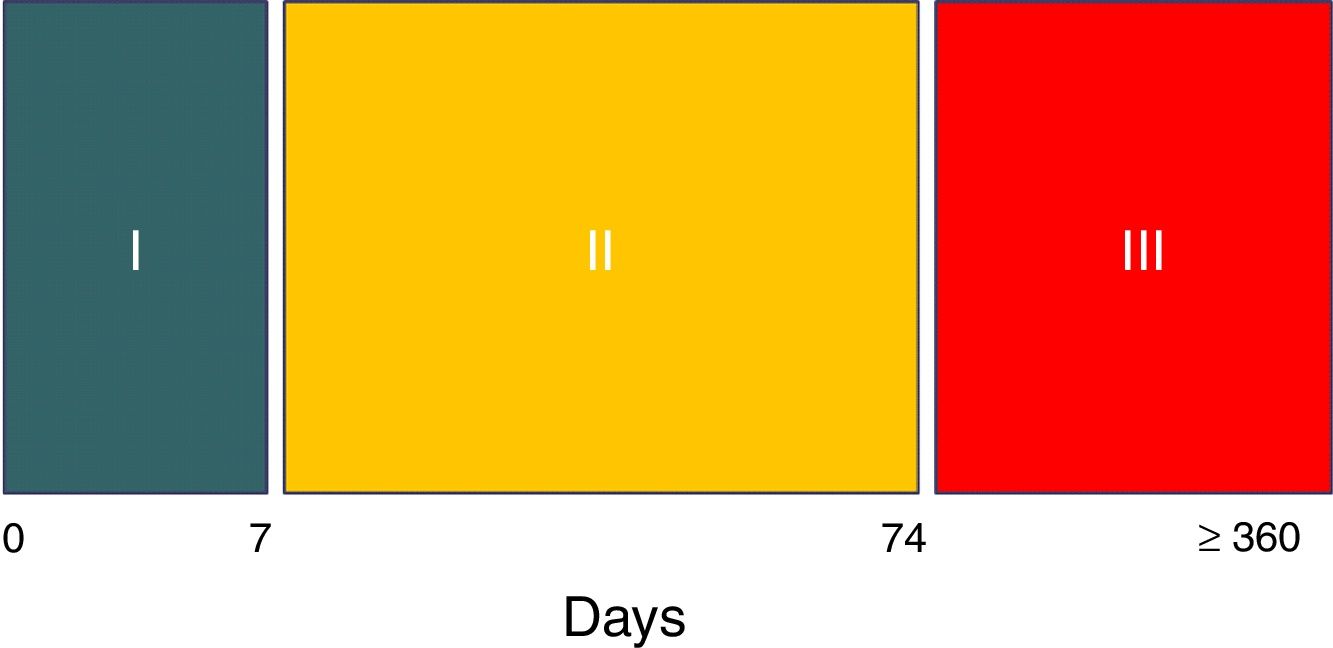
Evolutive stages of paroxysmal sympathetic hyperactivity.
Source: Hughes and Rabinstein.8
The reported incidence of PSH ranges from 8% to 33%,2–8 depending on the series and the underlying etiology of the disease. The disease can manifest in both children and adults,2–8 and has no particular age or gender predilection. A number of conditions predispose to the development of PSH, the most common being traumatic brain injury (80%) in its diffuse axonal damage presentation,9–14 followed by post-cardiac arrest anoxic-ischemic encephalopathy (10%) and cerebrovascular accident (CVA) (5.5%).2–8 In relation to CVA, spontaneous intraparenchymal bleeding of the basal nuclei, thalamus and cerebellar vermis, with or without ventricular collapse, is seen to predominate.15,16 There have also been reports of PSH in patients with severe subarachnoid hemorrhage,17,18 ischemic CVA, cerebral venous thrombosis, encephalitis and cerebral lipid embolism2–5,19,20 (Fig. 3).
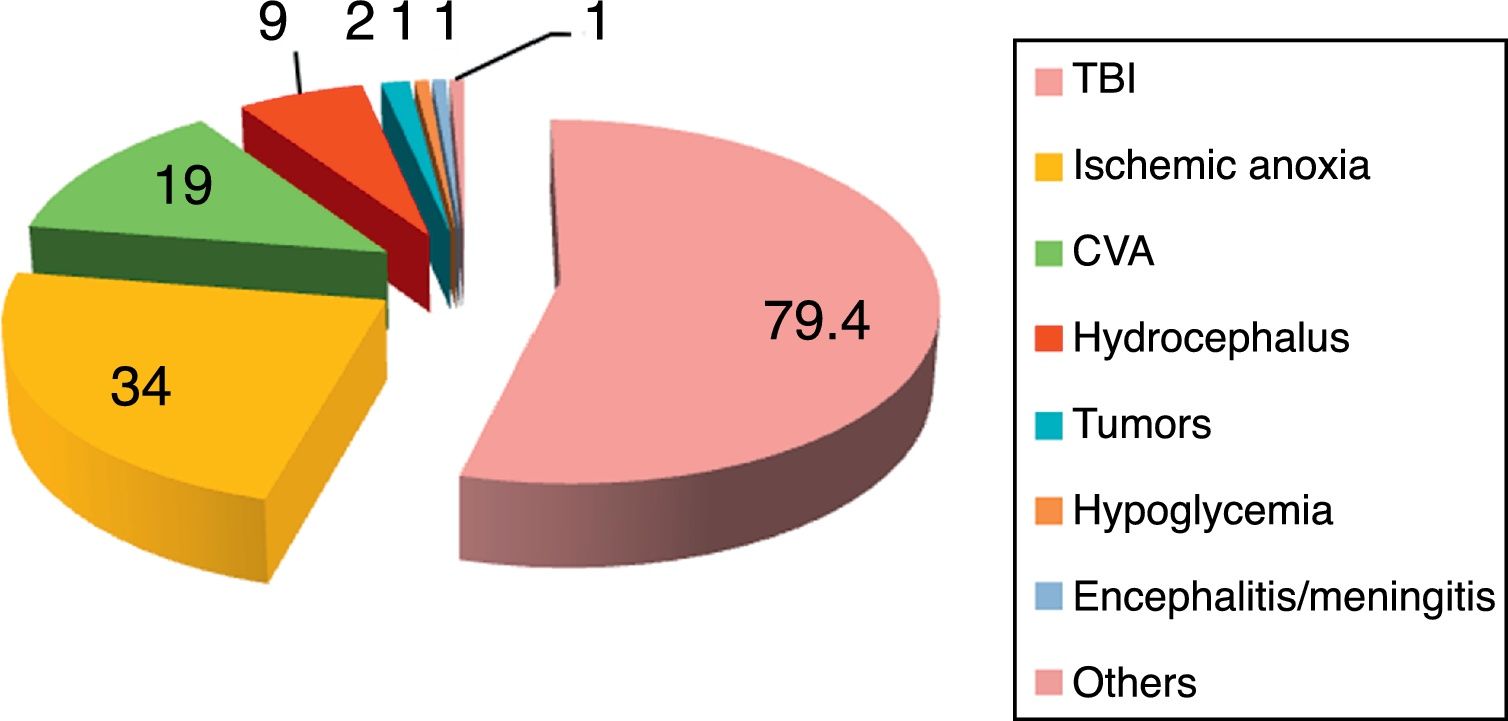
Disorders predisposing to the development of paroxysmal sympathetic hyperactivity.
CVA: cerebrovascular accident; TBI: traumatic brain injury.
Source: Perkes et al.2
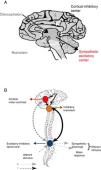
The main intervening element is the autonomic nervous system (ANS) or neurovegetative system, which is in charge of regulating involuntary actions through its three main components: sympathetic, parasympathetic and enteric.21 The ANS is essentially an efferent system. It receives information from the internal environment, glands and organs through a complex network located in the spinal cord, brainstem, diencephalon, hypothalamus, limbic system and certain areas of the brain cortex, and subsequently transmits impulses toward the periphery.21 In this way the ANS regulates heart rate, respiratory frequency, pupil diameter and blood vessel caliber, smooth muscle contraction, salivation, perspiration, sexual function, endocrine and exocrine gland secretion, and digestion.21
The sympathetic nervous system is composed of preaortic and pre- and paravertebral ganglionic networks. Catecholamines are the main intervening neurotransmitters, and are carefully regulated and balanced through excitatory and inhibitory impulses.21 When this balance is altered as a consequence of brain damage, catecholamine release becomes excessive, thereby giving rise to the clinical features typical of PSH.22,23
Why do imbalances resulting in catecholamine elevation during sympathetic paroxysm occur? The precise underlying physiopathology remains unclear. The syndrome was initially attributed to seizures or endocranial hypertension. Both conditions were discarded, however, since there is no close correlation between these two disorders and the presence of the syndrome.2–5,24,25 The absence of seizure activity as evidenced by the electroencephalogram (EEG) during sympathetic paroxysms has been clearly confirmed.2,24,25 On the other hand, a rise in intracranial pressure (ICP) is more a consequence than a cause of the syndrome.2–5
Following brain damage there is an immediate metabolic and inflammatory response, with activation of the ANS.26 This in turn induces tachycardia, arterial hypertension and the redistribution of blood flow toward the brain, heart and adrenal glands, in order to ensure the availability of oxygen and preserve the physiological functions of the vital organs of the body. The parasympathetic system in turn attempts to restore homeostasis by reducing the effects of sympathetic hyperactivity. However, when this parasympathetic feedback mechanism fails, sympathetic activity is disinhibited, hyperactivity results, and PSH ultimately develops. Two hypotheses have been proposed to explain this phenomenon24,25 (Fig. 4).
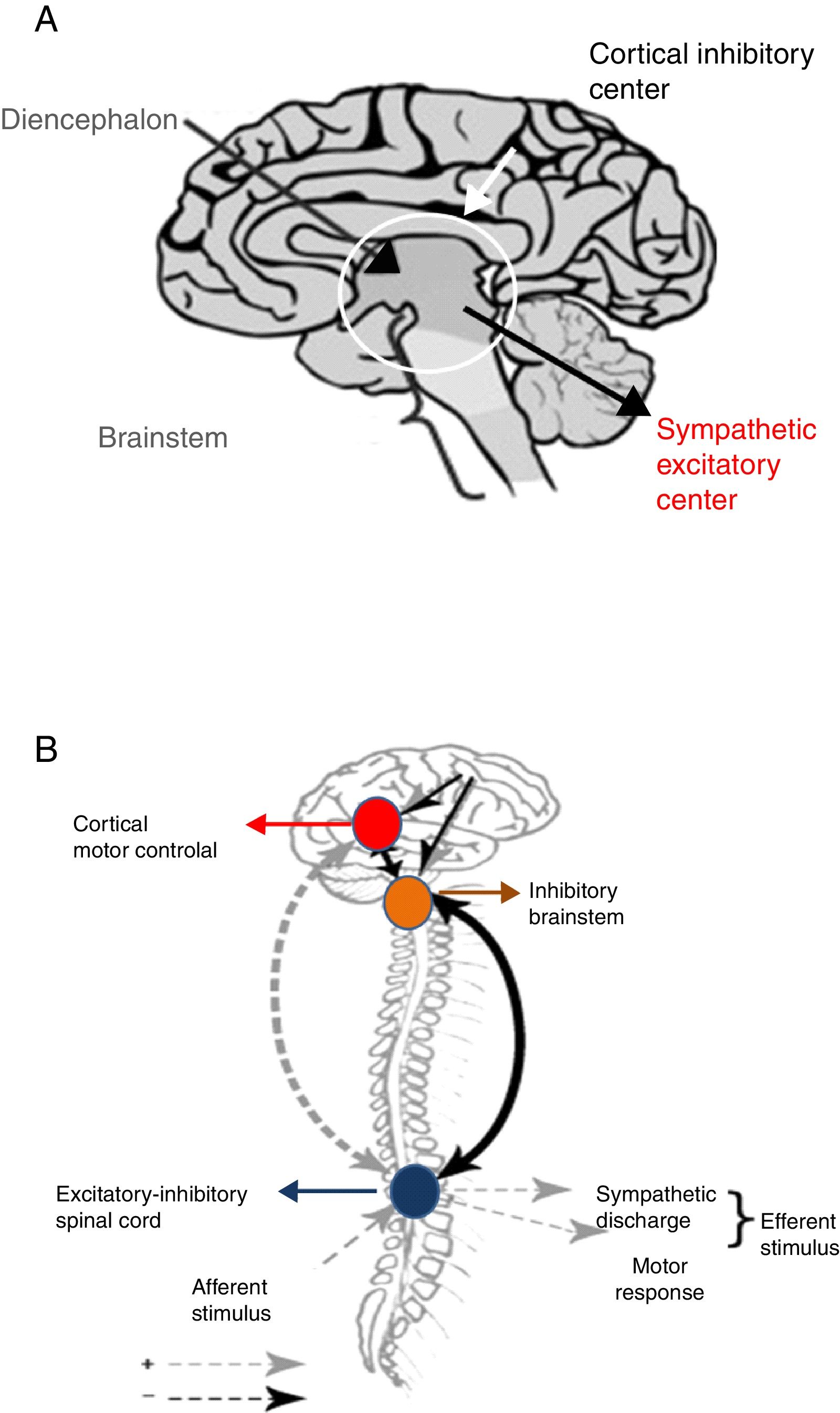
Disconnection theories: A. Conventional. B. Excitation-inhibition model.
Source: Baguley.25
The conventional theory of disconnection – whether structural (anatomical lesion) or functional (imbalanced neurotransmitter release) – postulates that sympathetic excitatory centers located in the diencephalon and upper region of the brainstem become freed from higher cortical-subcortical control.24
The excitation-inhibition ratio model in turn postulates that the centers located in the brainstem and diencephalon are inhibitory by nature. In this respect, they limit amplification and sensitization of the afferent sensory information coming from the spinal cord.24,25 In this model, PSH is referred to as allodynia – a term that defines a sensitization process occurring in the dorsal horn of the spinal cord, in which non-painful stimuli are perceived as painful, including the aspiration of secretions, body rotation, bathing, constipation and urinary retention.24,25 At the same time, painful stimuli become magnified in intensity.
On the other hand, the excitation-inhibition ratio model helps explain the triggering of PSH in response to environmental stimuli, as well as the response to drugs used to control the disorder.24,25
Question 5. How can a correct diagnosis be established?A strong degree of suspicion is required, particularly when in the context of acquired brain damage, during therapeutic de-escalation, “awakening from coma”, or in the rehabilitation phase, the patient suffers simultaneous and transient paroxysmal episodes of sympathetic hyperactivity.2,3,6–8,27
The predominant signs and symptoms are: tachycardia (98%), arterial hypertension (72%), excessive perspiration (79%), fever in the absence of an infectious focus (79%), tachypnea (85%), extensor body posture (38%), dystonia (38%), stiffness or spasticity (44%). Less frequent manifestations are: intermittent dilation of the pupils, diminished consciousness, hair erection, excitation and mechanical ventilator maladjustment.2,8
No complementary tests are available for confirming the diagnosis of PSH. The diagnosis is eminently clinical, and other situations involving similar clinical features therefore need to be ruled out2,3,6–8 (Table 2).
Disorders sharing signs or symptoms with paroxysmal sympathetic hyperactivity.
| Bacteremia, sepsis |
| Obstruction of the airway |
| Hypoxemia |
| Severe hypercapnia |
| Hypoglycemia |
| Seizures |
| Neurological deterioration (intracranial hypertension, bleeding, edema, hydrocephalus) |
| Pulmonary thromboembolism |
| Thyroid storm |
| Acute myocardial infarction |
| Alcohol or drug withdrawal |
| Sedation withdrawal syndrome, opioids |
| Malignant neuroleptic syndrome |
| Serotoninergic syndrome |
| Hyperthermia of central origin |
| Malignant hyperthermia |
Although the syndrome has been known for over 60 years, its diagnostic criteria are not homogeneous and have not been validated.2–8 Different numbers and combinations of signs have been used to confirm the existence of PSH,2,28 and only recently has expert consensus established its definition and the criteria to be used.29 Following a systematic review of the literature, a scale was developed based on the combination of a score that assesses the presence and severity of the clinical parameters (Clinical Features Severity [CFS]). This score is added to another score that assesses the characteristics of the episodes (frequency, duration, persistence over time, simultaneity, etc.), known as the Diagnosis Likelihood Tool (DLT)29,30 (Table 3). The final score obtained from the sum of these two scales allows us to calculate the probability of suffering PSH with increased precision, though its validation remains pending.
Diagnostic scale suggested by expert consensus.
| Clinical Features Severity scale (CFS) | |||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | Score | |
| HR | <100 | 100–119 | 120–139 | >140 | |
| RF | <18 | 18–23 | 24–29 | >30 | |
| SBP | <140 | 140–159 | 160–179 | >180 | |
| Temperature | <37 | 37–37.9 | 38–38.9 | >39 | |
| Perspiration | Null | Mild | Moderate | Severe | |
| Postures | Null | Mild | Moderate | Severe | |
| CSF subtotal | |||||
| Type and intensity of hypertonicity during the episode | ||
|---|---|---|
| Severity of the clinical presentation | Null | 0 |
| Mild | 1–6 | |
| Moderate | 7–12 | |
| Severe | >13 |
| Diagnosis Likelihood Tool (DLT) |
| Simultaneity of the clinical manifestations |
| Paroxysmal presentation |
| Sympathetic hyperactivity in response to non-painful stimuli |
| Persistence of the clinical manifestations ≥3 consecutive days |
| Persistence of the clinical manifestations ≥2 weeks post-injury |
| Persistence of the clinical manifestations despite treatment of alternative diagnoses |
| Treatment for reducing features of sympathetic hyperactivity |
| ≥2 daily episodes |
| Absence of parasympathetic clinical manifestations during the episode |
| Absence of other causes explaining the clinical manifestations |
| History of acquired brain damage |
| (1 point for each clinical presentation) DLT subtotal |
| Combination total (CFS+DLT) | ||
|---|---|---|
| Probability of diagnosis of PSH | Improbable | <8 |
| Possible | 8–16 | |
| Probable | >17 |
CFS: Clinical Features Severity scale; DLT: Diagnosis Likelihood Tool; PSH: paroxysmal sympathetic hyperactivity.
Source: Baguley et al.29
Although the catecholamine or other hormonal axis values for diagnosing the syndrome have not been defined, a recent study has generated the first proof of catecholamine elevation and, to a lesser extent, increased adrenocortical response, in individuals with PSH versus controls without the syndrome.23 These findings provide bases for warranting the current nomenclature referred to the disease.29
Question 6. Is neuroimaging necessary?Neuroimaging is not essential for diagnosing PSH. Nevertheless, it contributes to maintain a high level of suspicion by revealing the type of brain injury in the initial imaging study (focal or diffuse), its morphology (intracerebral hemorrhage, ischemia, contusions, extraaxial hematomas, etc.), the anatomical location, and the extent of the primary damage.31–33
Computed tomography (CT) is undoubtedly the imaging tool of choice in the acute phase. In more stable patients, without a need for multiple monitoring procedures, magnetic resonance imaging (MRI) with or without tractography can be used to more precisely define the degree of anatomical involvement – particularly as regards those anatomical structures more closely related to the development of PSH.34
Question 7. How should the syndrome be treated?Preventing PSH would be the ideal strategy. However, to date no measures in this regard have been successful. A recent retrospective study with important methodological limitations has indicated that the use of dexmedetomidine, versus the traditionally used sedatives (midazolam or propofol), in brain trauma patients subjected to surgery could reduce the incidence of PSH.35
As in any neurological emergency, the first aim of management is to ensure cardiorespiratory stability by means of the ABC of resuscitation in combination with the measures needed to detect and correct secondary lesions.2–5,36–39 Adequate nutrition and a correct water-electrolyte balance are very important assisting measures in patients with PSH.39
The existing drug strategies for the specific management of PSH are fundamented upon case series, most of which are retrospective and non-randomized. The quality of the evidence on the efficacy of the proposed measures is therefore poor. Likewise, it is important to note the lack of studies demonstrating the preference of one drug substance versus another. Nevertheless, the experience and the literature indicate that “drug combinations” are generally required, and that these should be evaluated through the so-called “trial and error” approach.3,5,20,36,38
In order to control excessive sympathetic activity or its consequences, it is important to orientate treatment according to the currently accepted physiopathology of PSH. In this regard, the paroxysms can be mitigated through three main approaches: (a) inhibition of central sympathetic flow; (b) inhibition of afferent sensory processes (preventing the development of allodynia); and (c) blockade of effector organ end response.3,5,20,36–38
At present, optimized treatment (effectiveness – adverse reactions – interactions) (Table 4) seeks the following:
- 1)
Abort crises or suspend symptoms: The aim here is to control the episode immediately in order to prevent adverse effects such as cardiac overload, arrhythmias, dehydration, muscle loss, contractures or delayed recovery from contributing to increase patient morbidity. The drugs used are characterized by rapid onset of action and a short half-life, and the choice of substance depends on the dominant symptom in each case. The indicated drugs include morphine, propranolol and short-acting benzodiazepines.3,5,20,36–38
- 2)
Prevention of paroxysms: Treatment in this regard seeks to reduce the frequency, duration and intensity of the symptoms, and is indicated in combination with the aforementioned therapy. The drugs used include nonselective beta-blockers (propranolol), α2-agonists (clonidine and dexmedetomidine in some groups of patients35), bromocriptine, baclofen, gabapentin and long-acting benzodiazepines.36,37
- 3)
Refractory PSH: When sympathetic hyperactivity persists despite the abovementioned treatments, with the risk of causing secondary brain damage, brain edema, lung edema, myocardial infarction, or catecholaminergic myocarditis including sudden death, use is made of drugs in continuous intravenous infusion, such as benzodiazepines, propofol, opioids or dexmedetomidine.38
Drugs used for the treatment of paroxysmal sympathetic hyperactivity.
| Drug | Mechanism | Mechanism of action | Starting dose | Frequency | Symptoms treated |
|---|---|---|---|---|---|
| Propranolol | Non-selective beta-blocker | Peripheral reduction of catecholamine effect | 40mg | Every 12h | Hypertension, tachycardia, fever |
| Morphine | Mu opioid agonist | Central and peripheral vagal modulation | 1–8mg | Conditioned to onset of PSH | Tachycardia, peripheral vasodilatation, allodynia response |
| Baclofen | Specific GABA agonist | Central | 5mg | Every 8h | Pain, clonus, stiffness |
| Gabapentin | GABA agonist | Central | 300mg | Every 8h | Spasticity, allodynia response |
| Benzodiazepines | GABA agonist | Central | Depends on the drug used | Agitation, hypertension, tachycardia, postures | |
| Bromocriptine | D2 dopaminergic agonist | Hypothalamic | 1.25mg | Every 12h | Dystonias, fever, postures |
| Clonidine | α2, Agonist | Reduces central sympathetic discharge | 0.1–0.3mg | Every 12h | Hypertension |
| Dexmedetomidine | α2, Agonist | Reduces central sympathetic discharge | 2μg/kg | Every 1h | Hypertension, agitation, tachycardia |
| Dantrolene | Reduces muscle contraction | Peripheral | 0.25–2mg/kg | Every 6–12h | Muscle stiffness, anomalous postures |
The effective clinical management of patients with PSH requires a clear understanding of the available treatment options, their efficacy, dosing characteristics, half-life, administration route, interactions and adverse effects. Treatment protocolization is therefore essential. A number of algorithms have been published in this respect.3,40
Since on one hand not all paroxysms are the same in terms of severity, frequency or duration, and on the other hand the available drug substances are not without potential toxic effects, we consider it important to categorize the episodes in order to use the treatment combination offering the best risk-benefit ratio. We thus recommend using the CFS+DLT diagnostic scale and classifying the patients into four possible groups (Fig. 5):
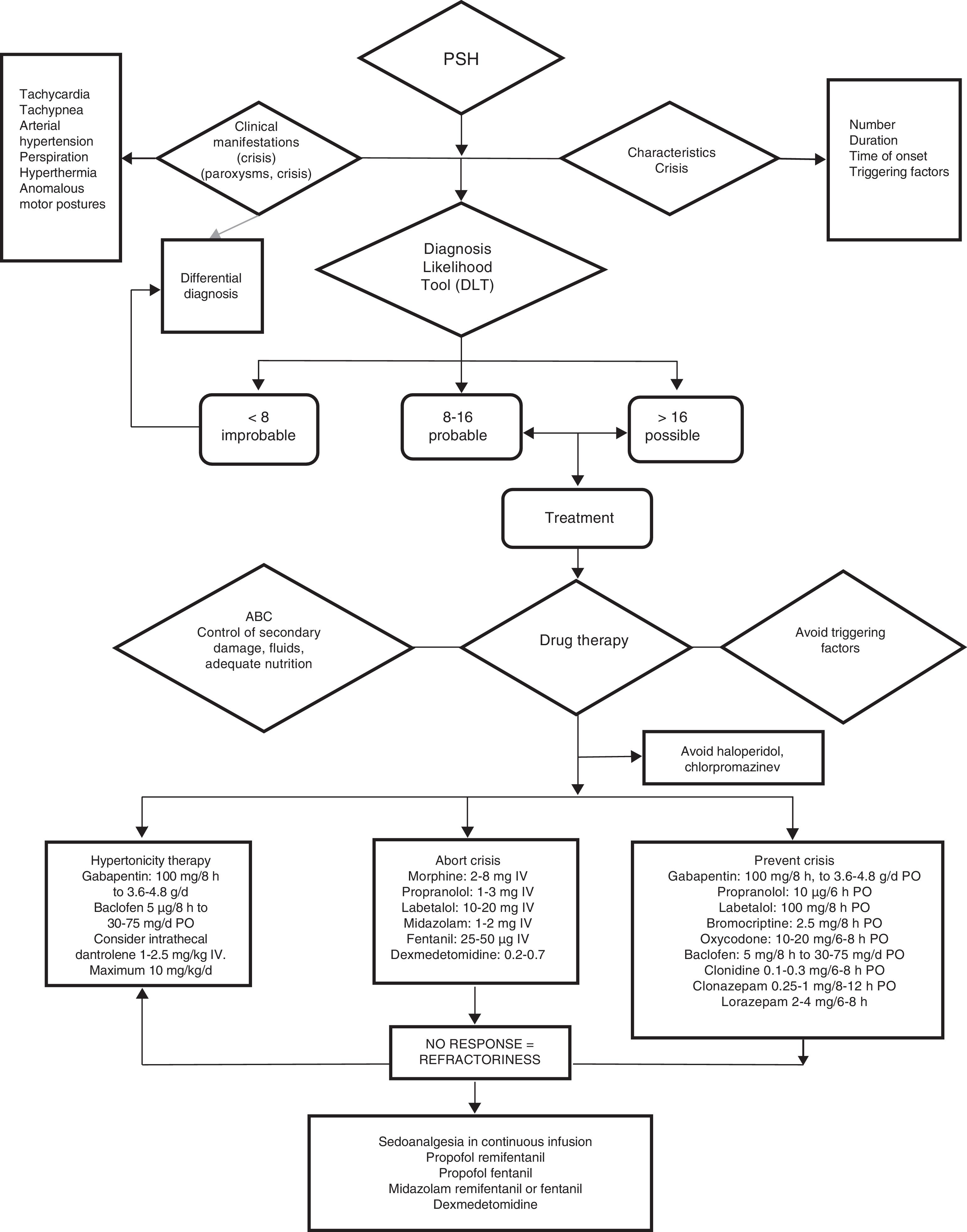
Management algorithm for paroxysmal sympathetic hyperactivity.
ABC of resuscitation: permeable airway, oxygenation and adequate ventilation, stable hemodynamic conditions.
Comment: haloperidol and chlorpromazine are to be avoided, due to their antidopaminergic effects that exacerbate or worsen PSH.
d: day; mg: milligrams; μg: micrograms; g: grams; IV: intravenous; PO: oral route.
Group A < 8 points. Treatment targeted to the dominant symptom.
Group B 8–16 points. Symptomatic+preventive treatment.
Group C > 17 points. Symptomatic+preventive treatment+gabapentin or dantrolene or baclofen.
Refractory group: Continuous intravenous infusion of propofol, fentanil, midazolam or dexmedetomidine.
Question 8. What are the consequences of a lack of diagnosis or adequate management?Paroxysmal sympathetic hyperactivity episodes can be intense and prolonged, and can occur several times a day.2–6,41 The number of symptoms rather than the duration of PSH is the most important severity indicator.4 Arterial hypertension, fever, hypoxemia, hypercapnia and hyperglycemia can cause secondary brain damage, and are the main causes of a poor prognosis.2–6 Paroxysmal sympathetic hyperactivity in turn induces a hypermetabolic state, with hypercatabolism and inflammation, and increased vulnerability to infections, sepsis and weight loss, which in turn are associated to increased morbidity, longer hospital stay and slower recovery.2–6,20 The marked and sustained increase in catecholamine levels predisposes to the development of myocardiopathy, lung edema, arrhythmias, and cardiac and multisystemic dysfunction.2–8,20 An early diagnosis and optimized treatment of PSH are crucial in order to facilitate patient recovery and avoid permanent disabilities secondary to heterotopic ossification, spastic stiffness, body malpositioning and profound neurocognitive disturbances.2–8,20 The early start of specific symptoms therapy is believed to reduce the complications rate, shorten stay in the Intensive Care Unit (ICU) and facilitate recovery.18
Diagnostic and/or therapeutic delays in PSH can have devastating consequences for patient recovery. However, on comparing the post-rehabilitation course of patients with and without PSH, functional condition at discharge shows no statistically significant differences as determined from the Functional Independence Measure, Disability Rating Scale and Glasgow Outcome Scale – though the probability and degree of recovery is greater in those without PSH.42,43
The expectable course of recovery from traumatic brain damage extends from acute management to rehabilitation and reinsertion in the community. The effects of PSH can alter the course of rehabilitation and delay or impede maximum patient recovery.43
Conclusions and future challengesParoxysmal sympathetic hyperactivity is a genuine and potentially fatal neurological emergency. The diagnosis is based on the clinical findings; as a result, the professionals in charge of the care of neurocritical patients must be able to detect the syndrome and require adequate training. The greatest challenge in current routine practice is to ensure the early identification of PSH, and over the short term this requires homogenization and validation of both the nomenclature used and the diagnostic criteria employed – thereby facilitating comparison of the efficacy of future treatments. Early categorization is essential (not all paroxysms share the same clinical features or characteristics), in the same way as work on a systematic and multidisciplinary basis. The protocolization of treatment is crucial, and should be scaled exclusively according to the patient clinical picture. The aim is to ensure early and intensive care in order to prevent complications and optimize the chances for rehabilitation.
Conflicts of interestNone.
Please cite this article as: Godoy DA, Panhke P, Guerrero Suarez PD, Murillo-Cabezas F. Hiperactividad simpática paroxística: una entidad que no debería pasar desapercibida. Med Intensiva. 2019;43:35–43.

