












Editado por: F. Baigorri-González y J.º. Lorente Balanza
Última actualización: Abril 2005
Más datosFISIOLOGÍA DE LA COAGULACIÓN
Casi 40 años han transcurrido desde la primera descripción del modelo clásico de cascada de la coagulación que establecía dos vías (intrínseca y extrínseca) que generaban factor Xa y finalmente trombina. Este modelo sirve para explicar las pruebas clínicas de coagulación más empleadas: tiempo de protrombina (vía extrínseca) y tiempo parcial activado de tromboplastina (vía intrínseca). Actualmente se ha propuesto un modelo celular que destaca como proceso clave de la hemostasia in vivo la formación del complejo factor tisular-VIIa (FT-VIIa) y que explica algunas inconsistencias clínicas de la cascada de la coagulación. El factor tisular (tromboplastina) es un receptor transmembrana para el factor VII siendo similar a los receptores de las citocinas. Tras la formación del complejo FT-VIIa se genera factor Xa, IXa y trombina en la superficie de las células portadoras de FT. La acumulación en la superficie de la plaqueta activada de los cofactores activados y su unión a los factores activados da lugar a trombina y la polimerización ulterior de la fibrina. Así, la hemostasia in vivo abarca dos procesos, ambos iniciados por el complejo FT-VIIa, en el primero se genera Xa en la célula portadora de FT y proporciona pequeñas cantidades de trombina a partir de la protrombina, activa la plaqueta, libera el factor VIII del factor von Willebrand y activa el factor V en la superficie plaquetaria. En el segundo se produce factor IXa que al unirse con el VIIIa en la superficie de la plaqueta activada cataliza la formación de Xa y gran cantidad de trombina en la superficie plaquetaria.
En la figura 1 se representa un esquema actual de la coagulación. La formación del complejo prekalikreina (Pk), kininógeno de alto peso molecular (Ki) y factor XI y XII en contacto con el colágeno subendotelial genera finalmente el factor XIa (vía intrínseca o de contacto). El FT expresado en el endotelio vascular une y activa el factor VII (vía extrínseca) siendo éste el mecanismo fundamental de iniciación de la coagulación in vivo. El complejo de factores IX, X y el cofactor VIIIa en la superficie endotelial o plaquetaria permite la activación del factor IX que a su vez activa el factor X. El factor Xa en presencia del cofactor V permite el paso de protrombina (PT) a trombina (T). La generación de trombina es mucho mayor en la superficie plaquetaria. Las uniones de los factores VIIa, IX, X, Xa y PT a la superficie celular se realizan por la presencia de complejos de calcio, fosfolípido y ácido dicarboxiglutámico, siendo este último paso consecuencia de la acción de la vitamina K sobre los factores dependientes de la misma. La formación de trombina actúa como un mecanismo amplificador ya que favorece la activación de plaquetas y la activación del factor XI y del cofactor VIII y V (fig. 2). Además se une a los receptores de trombina, acoplados a proteínas G, presentes en el endotelio, linfocitos, monocitos, macrófagos y neutrófilos, manteniendo así la hemostasia a nivel local y proporcionando un mecanismo de activación inflamatoria.
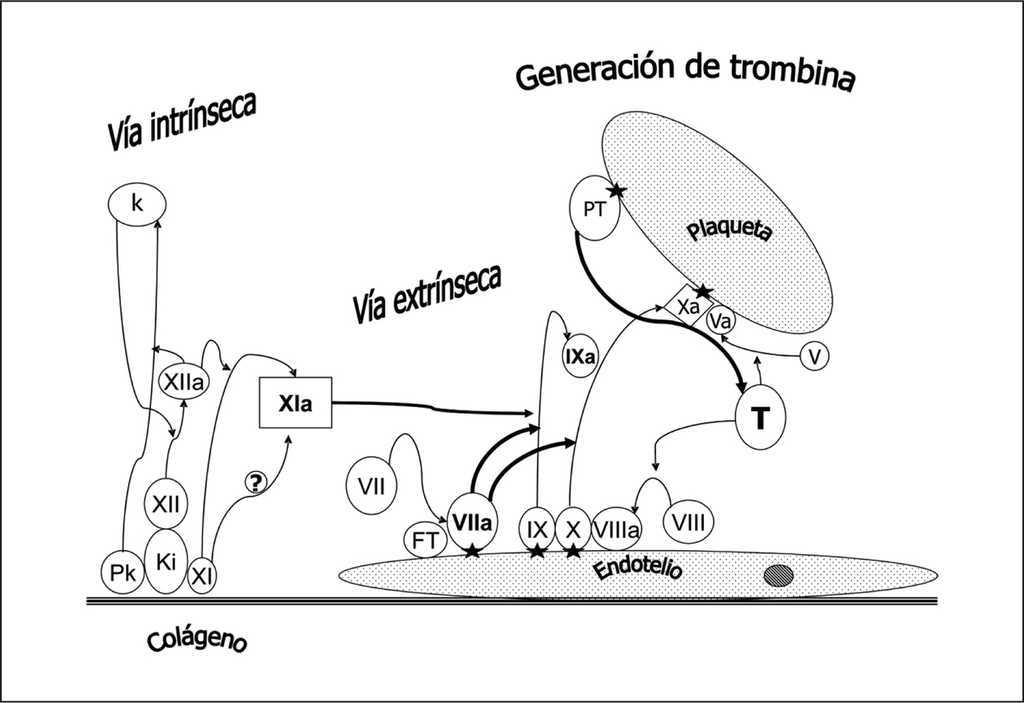
Figura 1. Esquema de la coagulación. La formación del complejo prekalikreína (P), kininógeno de alto peso molecular (Ki) y factor XI y XII en contacto con el colágeno subendotelial genera finalmente el factor XIa (vía intrínseca o de contacto). El factor tisular (FT) expresado en el endotelio vascular une y activa el factor VII (vía extrínseca), siendo éste el mecanismo fundamental de iniciación de la coagulación in vivo. El complejo de factores IX, X y el cofactor VIIIa en la superficie endotelial o plaquetaria permite la activación del factor IX que a su vez activa el factor X. El factor Xa en presencia del cofactor V permite el paso de protrombina (PT) a trombina (T). La generación de trombina es mucho mayor en la superficie plaquetaria. Las uniones de los factores VIIa, IX, X, Xa y PT a la superficie celular se realizan por la presencia de complejos de calcio, fosfolípido y ácido dicarboxiglutámico (estrella). Se han representado separadamente los diferentes pasos entre el endotelio y la plaqueta para facilitar su comprensión aunque en la realidad dista de existir tal separación.
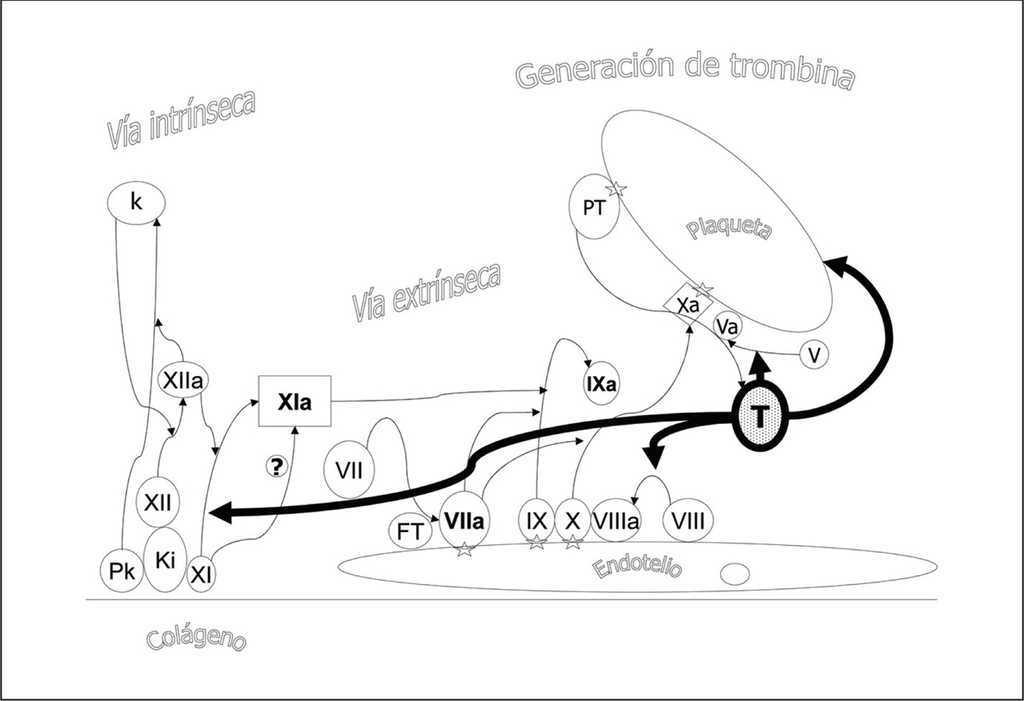
Figura 2. Efectos de la trombina. La trombina actúa amplificando la cascada de la coagulación fomentando la activación plaquetaria, y la activación del factor XI y los cofactores V y VIII.
La acción de la trombina sobre el fibrinógeno circulante genera monómeros de fibrina y fibrinopéptidos A y B. Posteriormente se polimeriza la fibrina y por acción de la trombina que activa el factor XIII se entrecruzan las fibras de fibrina dando lugar a la fibrina estable que estructura el coágulo plaquetario. Inmediatamente se pone en marcha un mecanismo para la lisis del coágulo: el sistema fibrinolítico endógeno (fig. 3). A partir del plasminógeno (P) embebido en el coágulo y por acción del activador del plasminógeno tisular (tPA), sobre todo, y en menor medida por el activador del plasminógeno urinario (o urokinasa, UK) ambos producidos por la célula endotelial vascular, se genera plasmina (p) que degrada la fibrina estable a productos de degradación como el dímero D. La estreptokinasa (SK) producida por el estreptococo actúa de modo similar. La plasmina que escapa del coágulo al plasma se inactiva rápidamente por la α2 plasmina inhibidor (α2PI) limitando así su acción al coágulo local. La acción profibrinolítica del tPA está a su vez controlada por la acción de dos inhibidores cuya formación está estimulada por la trombina que contribuye así a modular una fibrinólisis incontrolada: el inhibidor endotelial del activador del plasminógeno (PAI) y el inhibidor de la fibrinólisis activado por trombina (TAFI). Por otro lado, la bradikinina (BK) generada a partir de Ki por acción del factor XIIa, actúa sobre el plasminógeno formando plasmina.
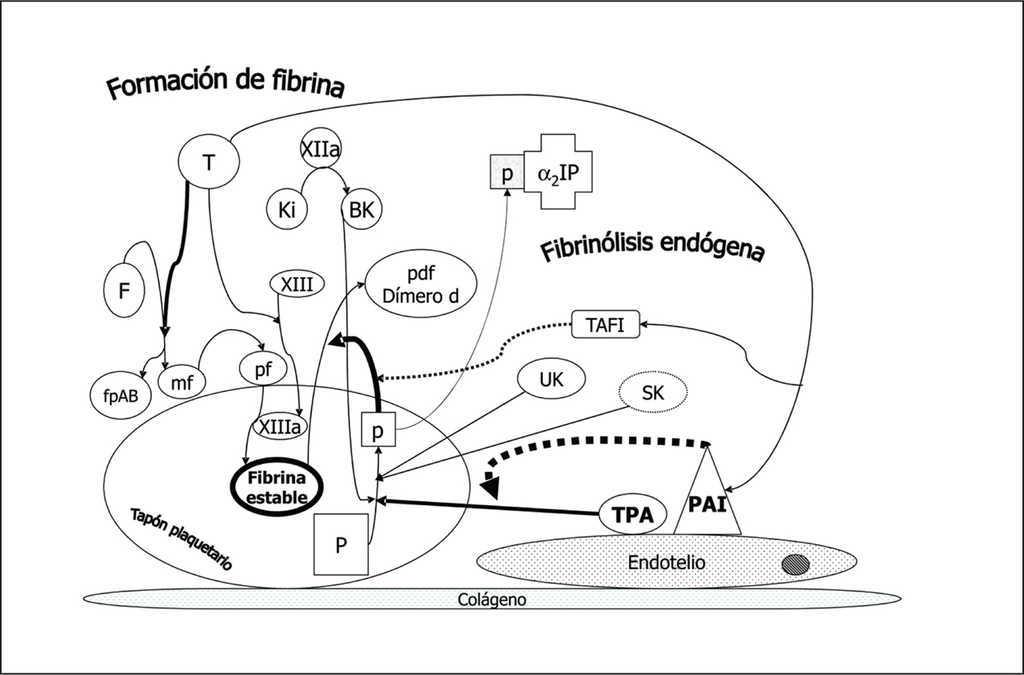
Figura 3. Fibrinólisis endógena. El fibrinógeno (F) por acción de la trombina (T) forma monómeros de fibrina (mf) liberando fibrinopéptidos A y B con capacidad procoagulante. Los mf se polimerizan (pf) y éstos por acción del factor XIIIa (activado por acción de la T) constituyen la fibrina estable con sus fibras entrecruzadas. El plasminógeno (P) embebido en el coágulo de plaquetas y fibrina estable se transforma en plasmina (p) por la acción de: 1. la bradikinina (BK) generada a partir del kininógeno de alto peso molecular (Ki) por acción del factor XIIa; 2. el activador del plasminógeno urinario (UK) que actúa sobre todo en el plasminógeno circulante, y 3. el activador del plasminógeno tisular (tPA), activador fisiológico primario. La estreptokinasa (SK) producida por el estreptococo actúa de un modo similar. La p transforma la fibrina estable en productos de degradación como el dímero d, y la que escapa a la circulación queda ligada y neutralizada por el alfa 2 plasmina inhibidor (alfa2IP). Dos inhibidores endógenos de la fibrinólisis, bajo control de la trombina, el inhibidor endotelial del activador tisular (PAI) y el inhibidor de la fibrinólisis activable por trombina (TAFI) actúan inhibiendo la acción del tPA y de la formación de p, respectivamente.
El sistema de la coagulación a través del factor XII (Hageman) interacciona con el sistema de kininas, el sistema del complemento y el sistema fibrinolítico (fig. 4). El factor XII activado genera bradikinina a partir del kininógeno que a su vez favorece la formación de plasmina a partir del plasminógeno y ésta, la activación del factor C3. Por otro lado el factor XII activado libera a partir de la prekalikreína, kalikreína que amplifica la activación del factor XII y activa el factor C5.
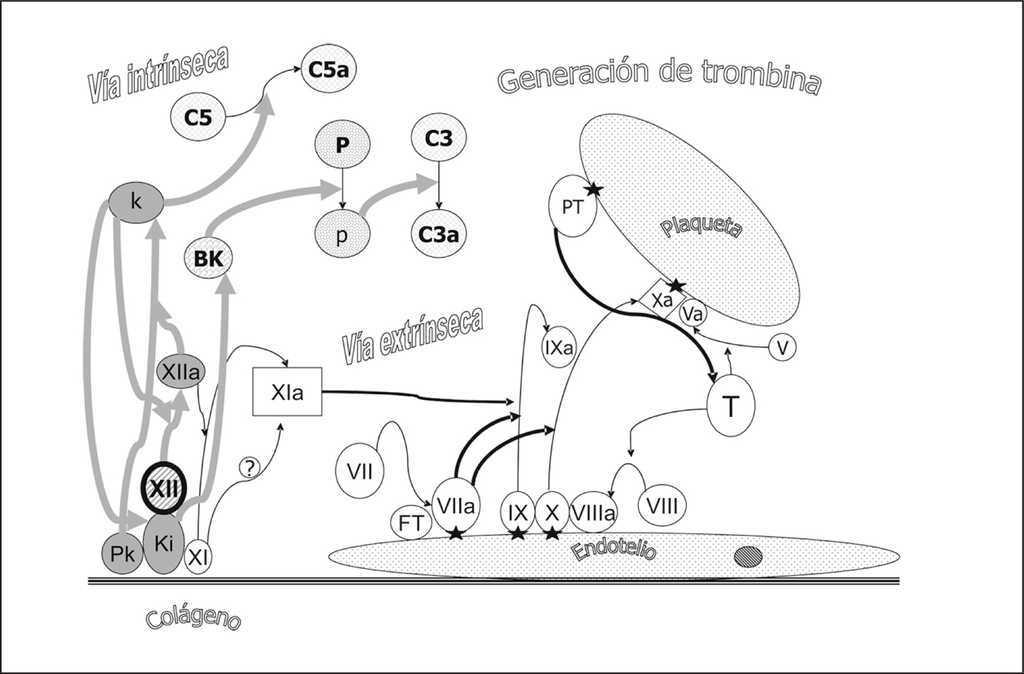
Figura 4. Acciones del factor XIIa (Hageman) en la interacción del sistema de coagulación y los sistemas de cininas, fibrinolítico y del complemento. La activación del factor XII conlleva la formación de kalikreína (k) a partir de prekalikreína (PK) activando el factor C5, y la liberación de bradikinina (BK) del kininógeno de alto peso molecular (Ki) fomenta la formación de plasmina (p) a partir de plasminógeno (P). La plasmina activa el factor C3 a C3a.
El potencial procoagulante de 1 ml de sangre es suficiente para coagular todo el sistema circulatorio si no fuera por la existencia de mecanismos anticoagulantes que, con un control extremadamente fino, mantienen el equilibrio de la coagulación para que se mantenga a nivel local donde se ha producido la lesión y no se propague. Este sistema anticoagulante endógeno incluye la antitrombina, el inhibidor de la vía del factor tisular (TFPI) y el sistema de la proteína C.
La antitrombina (AT) tiene un peso molecular de 58.200 dalton con 432 aminoácidos, es producida en el hígado y su concentración plasmática es de 112 a 140 mg/l con una vida media de dos a tres días. Es un regulador importante de la cascada de la coagulación en su función de serpina (inhibidor de las serina proteasas) de la coagulación. En la primera mitad del siglo XX se describieron diferentes actividades de antitrombina en el plasma dando lugar a la clasificación de las antitrombinas (I a IV). Posteriormente se demostró que las diferentes actividades eran fruto de sólo una molécula, la antitrombina III, nombre que se acordó simplificar como antitrombina en 1993 por la Sociedad Internacional de Trombosis y Hemostasis1. La AT es un cofactor de la heparina actuando como un inhibidor fisiológico de la trombina (T) y factor Xa (fig. 5). En menor medida inhibe los factores IXa, XIa, XIIa, kalikreína y plasmina. Su acción se multiplica por mil en presencia de heparina. Su activación fisiológica se produce por el heparán sulfato ligado a riuducan o sindecán en la superficie endotelial.
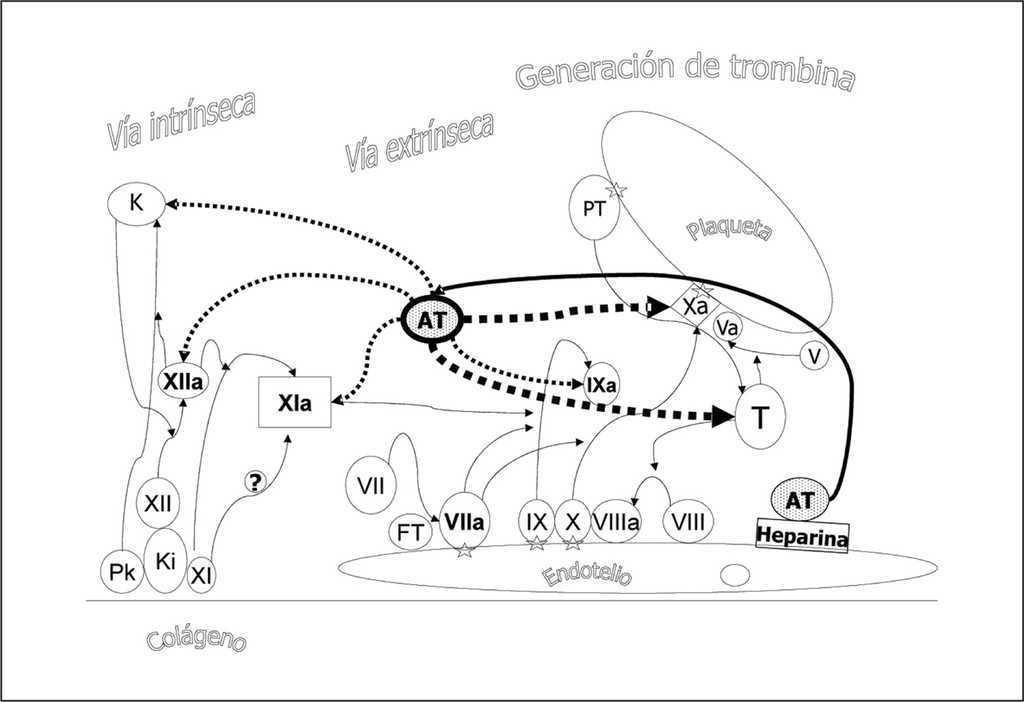
Figura 5. Efectos de la antitrombina. La antitrombina (AT) es un cofactor de la heparina y un inhibidor de las serinaproteasas (serpina) que intervienen en la coagulación, actuando como un inhibidor fisiológico de la trombina (T) y factor Xa. En menor medida inhibe los factores IXa, XIa, XIIa, kalikreína y plasmina. Su acción se multiplica por mil en presencia de heparina. Su activación fisiológica se produce por el heparán sulfato ligado a riuducán o sindecán en la superficie endotelial.
El TFPI es una serpina producida por las células endoteliales vasculares encontrándose ligado a glucosaminoglicanos de la membrana y que se libera a la circulación, ligado a lipoproteínas, en presencia de heparina2. En las plaquetas se almacena una pequeña cantidad que se libera cuando se activan. El TFPI inhibe directamente el factor Xa y dependiendo de éste el complejo catalítico FT-VIIa (fig. 6).

Figura 6. Acción anticoagulante del inhibidor de la vía del factor tisular (TFPI). El TFPI es producido por la célula endotelial y actúa formando un complejo cuaternario con el factor tisular y los factores VIIa y Xa. Su acción global es inhibir la acción activadora del factor VIIa en los factores IX y X.
La proteína C (PC) es una serina proteasa sintetizada en el hígado y dependiente de vitamina K, con un peso molecular de 55.000 dalton. Las concentraciones plasmáticas de PC (4 ug/ml) son de 2.000 a 4.000 veces las de su forma activada (PCA, 1 a 2 ng/ml). La PC plasmática es heterogénea con diferentes variantes estructurales que dependen de modificaciones en la cadena ligera. Los niveles de PC alcanzan el límite inferior de la normalidad a los 6-12 meses de edad. Esta maduración tardía del sistema de la PCA explicaría que las deficiencias de proteína C en la sepsis pediátrica sean más pronunciadas que en el adulto. En situaciones normales el sistema de la proteína C desempeña un papel crítico para el mantenimiento de la hemostasis inhibiendo la conversión de protrombina a trombina por su efecto inhibidor sobre los cofactores Va y VIIIa (fig. 7). La PC ligada al receptor endotelial (REPC) y cuando la trombina (T) se ha unido al complejo de trombomodulina (TM) endotelial se activa a PCA que en presencia de la proteína S (S) inhibe los cofactores Va y VIIIa. Por otro lado, la PCA tiene un efecto profibrinolítico por dos mecanismos: actúa inhibiendo el efecto del PAI sobre la acción del tPA y además inhibe el efecto activador de la trombina sobre el inhibidor de la fibrinólisis activable por trombina (TAFI) disminuyendo así su efecto antifibrinolítico (fig. 8).
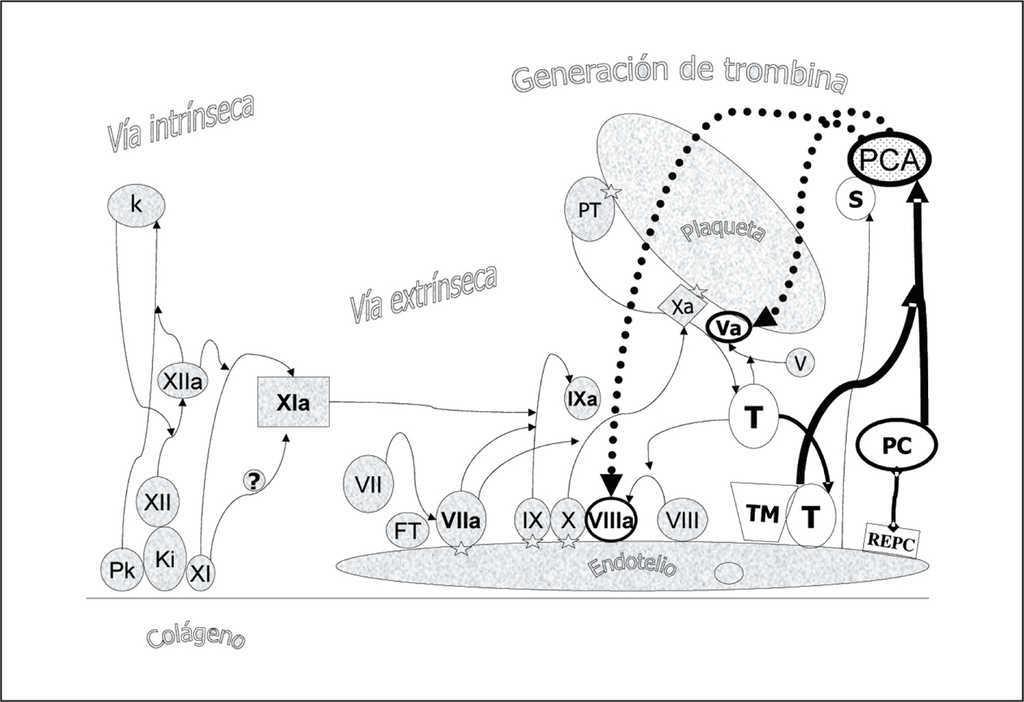
Figura 7. Efecto anticoagulante de la proteína C (PC). La PC ligada al receptor endotelial y cuando la trombina (T) se ha unido al complejo de trombomodulina (TM) endotelial se activa a proteína C activada (PCA) que en presencia de la proteína S (S) inhibe los cofactores Va y VIIIa.
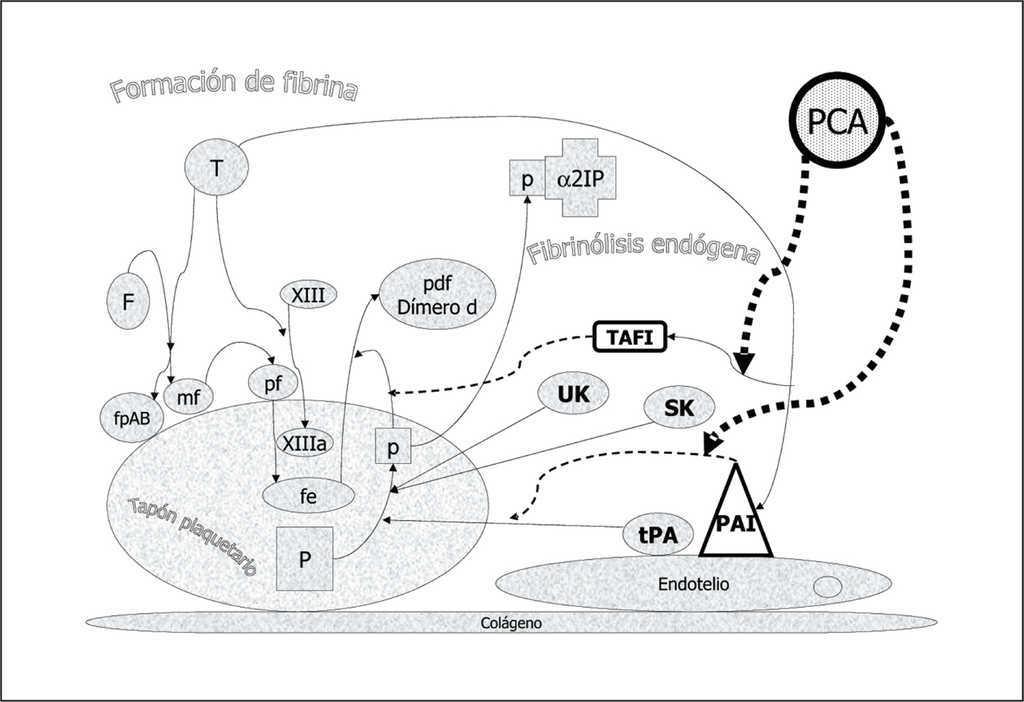
Figura 8. Efecto profibrinolítico de la proteína C activada (PCA). La PCA actúa inhibiendo el efecto del inhibidor del activador del plasminógeno (PAI) sobre la acción del activador tisular del plasminógeno (tPA). Además inhibe el efecto activador de la trombina (T) sobre el inhibidor de la fibrinólisis activable por trombina (TAFI) disminuyendo así su efecto antifibrinolítico.
COAGULOPATÍA DE LA SEPSIS
La hipótesis que exploramos considera la sepsis grave como un síndrome caracterizado por un estado procoagulante, proinflamatorio y antifibrinolítico en el que múltiples estirpes celulares con potencial inflamatorio incluyendo monocitos, macrófagos, neutrófilos y endotelio vascular, liberan citocinas diversas como interleucina 1 (IL-1), factor de necrosis tumoral alfa (TNF-alfa) y otros mediadores inflamatorios. Éstos, a su vez, dañan el endotelio que expresa el factor tisular y se activa así la vía extrínseca de la coagulación generando trombina y finalmente fibrina que se deposita en la microcirculación. Ésta microtrombosis generalizada induciría una lesión de isquemia-reperfusión en diferentes órganos. Ésta es una de las hipótesis para explicar la disfunción multiorgánica que conduce a la muerte en más del 40% de los pacientes con sepsis grave.
Hace más de 30 años que se correlacionó la activación de la coagulación con el shock en pacientes sépticos independientemente del tipo de germen. En los pacientes con sepsis se observan frecuentemente alteraciones de la coagulación y fibrinólisis que probablemente están relacionadas con la presencia de mediadores inflamatorios y los cambios del endotelio vascular inducidos por la sepsis3,4. Se ha demostrado la formación de microtrombos en la microcirculación hepática en respuesta a la administración de endotoxina dando lugar a hipoperfusión, necrosis tisular y disfunción multiorgánica5. La coagulopatía microvascular de la sepsis se manifiesta en la frecuente presencia de trombocitopenia y niveles elevados de productos de degradación de la fibrina o dímeros D y esto, a su vez, se correlaciona con la mortalidad6. Aunque menos del 20% de los pacientes con sepsis grave presentan coagulación intravascular diseminada manifiesta, casi todos tienen concentraciones elevadas de dímero-D y alteraciones de la proteína C7.
En los modelos experimentales de sepsis se comprueba la existencia de cambios microscópicos compatibles con daño celular en las células endoteliales vasculares a los pocos minutos de inyectar lipopolisacárido bacteriano (LPS). Durante las primeras 24 horas se hacen patentes cambios globales que conllevan cambios en la permeabilidad vascular facilitando el paso de líquido y células inflamatorias al espacio intersticial. Se han detectado células endoteliales circulantes, desprendidas del endotelio vascular dañado, en la sepsis y el shock séptico. Igualmente aumenta la expresión del factor tisular en el endotelio y en células mononucleares desencadenando la vía extrínseca y contribuyendo a favorecer un estado de coagulación generalizada (fig. 9). Por otro lado, se expresa a nivel endotelial menos TM (aunque aumentan los niveles plasmáticos de TM inactiva), menos REPC, menos heparan sulfato (H) y menos TFPI. Todos estos cambios inhibidores de anticoagulantes naturales potencian el efecto procoagulante inicial del FT.
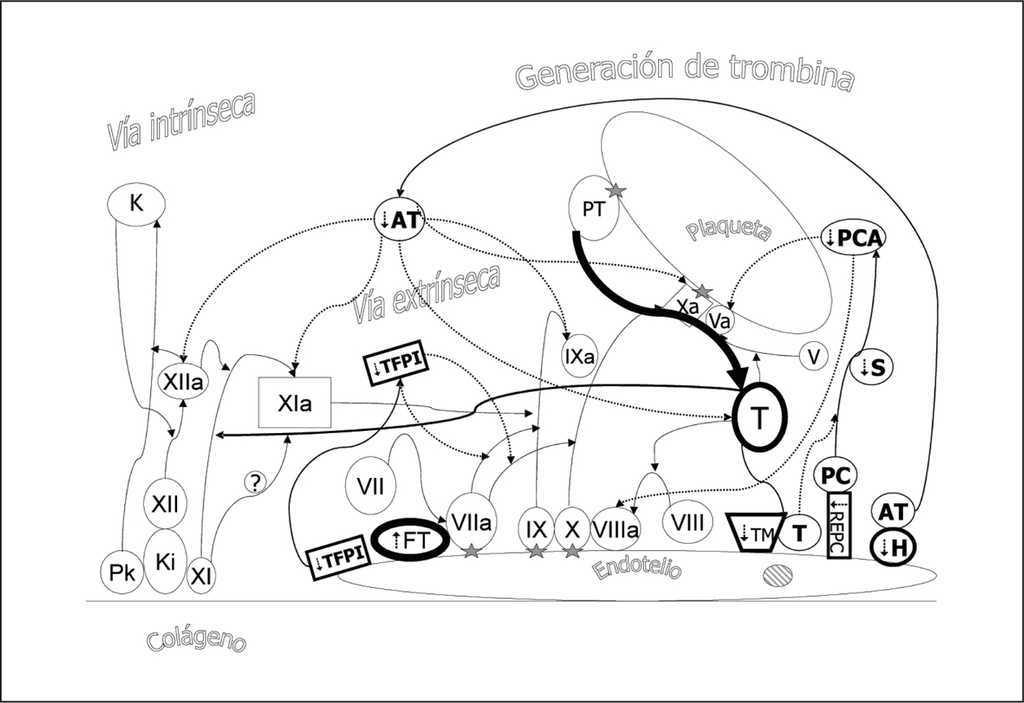
Figura 9. Efecto procoagulante de la sepsis. En la sepsis se produce una mayor expresión del factor tisular (FT) en el endotelio vascular y en células mononucleares desencadenando la vía extrínseca. Por otro lado se expresa a nivel endotelial menos trombomodulina (TM), menos receptor endotelial de proteína C (REPC), menos heparán sulfato (H) y menos inhibidor de la vía del factor tisular (TFPI). Todos estos cambios inhibidores de anticoagulantes naturales potencian el efecto procoagulante inicial del FT.
El descenso de TM que inhibe los efectos de la trombina para amplificar la coagulación y actuar sobre los receptores inflamatorios de trombina conlleva también la incapacidad de la trombina para activar la PC. El descenso observado de PCA en pacientes con sepsis contribuye a perpetuar el mecanismo de coagulación por la pérdida del efecto inhibidor de la PCA en los cofactores V y VIII y en la acción del PAI y del TAFI sobre la fibrinólisis. Por otro lado, se observa una disminución de la expresión endotelial del REPC. El endotelio expresa menos moléculas de heparina endógena (H), potenciadora de la AT, perdiendo ésta su capacidad para inhibir los factores de coagulación. En la sepsis, el endotelio libera menos tPA y aumenta la producción de su inhibidor (PAI), cambios globales que frenan la fibrinólisis endógena (fig. 10).
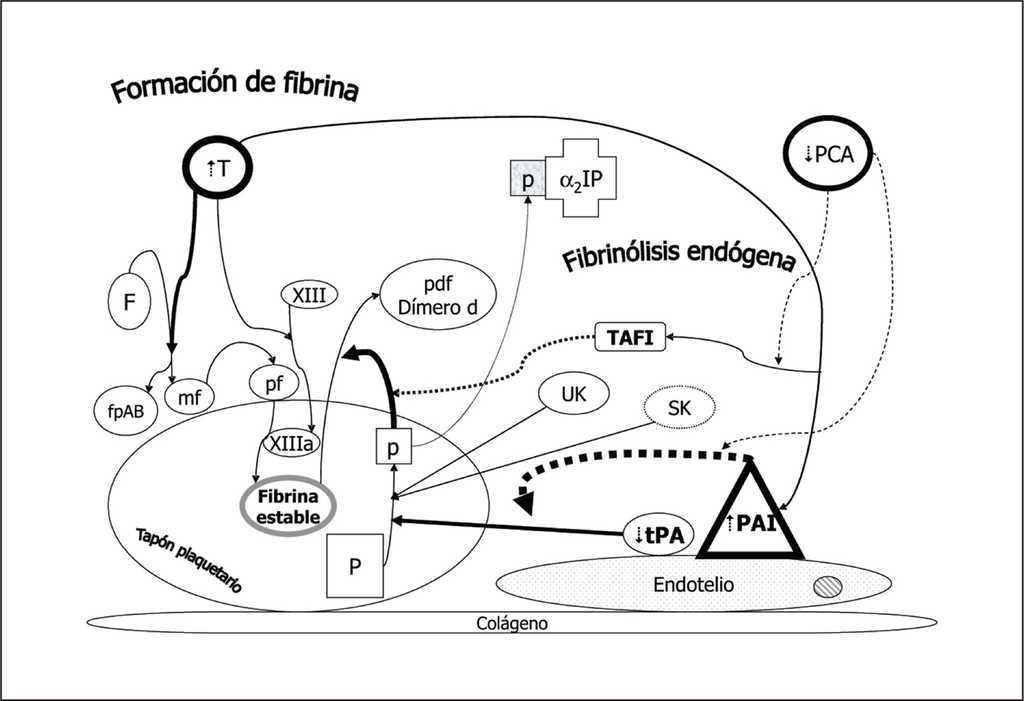
Figura 10. Efecto antifibrinolítico de la sepsis. En la sepsis se observa una disminución en la expresión endotelial del activador del plasminógeno tisular (tPA) y urinario (UK) junto con el aumento de la producción del inhibidor del activador del plasminógeno (PAI). Por otro lado la trombina producida en la coagulopatía de la sepsis aumenta el efecto del PAI y del inhibidor de la fibrinólisis activable por trombina (TAFI). La disminución en la producción de proteína C activada (PCA) que debería inhibir los efectos del PAI y TAFI, hace que el resultado final sea antifibrinolítico.
La sepsis induce cambios específicos diferentes en el endotelio vascular de los distintos órganos explicando las diferencias en respuesta de los mismos a la estimulación inflamatoria global de la sepsis. Estos cambios inflamatorios podrían ser distintos según la causa de la sepsis y las diferencias genéticas individuales contribuyendo a la variada expresividad clínica del síndrome8.
ANTITROMBINA EN LA SEPSIS GRAVE
En 1965 se describió la primera asociación genética entre la deficiencia de un anticoagulante natural, la AT, y la trombosis venosa. En 1981 y 1984 se describieron las deficiencias de proteína C y S respectivamente. Estudios animales sugerían que la antitrombina III (AT) en dosis suprafisiológicas tenía un efecto antiinflamatorio al unirse a ciertos glucosaminoglicanos endoteliales. El resultado es un aumento en la producción de prostaciclina endotelial que limita la interacción con neutrófilos y plaquetas disminuyendo la producción de citocinas. Curiosamente este efecto es abolido por la heparina, ya que inhibe competitivamente la unión a los glucosaminoglicanos desviando su efecto en el sentido anticoagulante (fig. 5). En la sepsis disminuye la síntesis hepática de AT y se degrada más por la elastasa neutrófila. Múltiples estudios mostraban una disminución de AT en la sepsis y sugerían que podía proteger de la disfunción multiorgánica (DMO) y disminuir la mortalidad. Se llevó a cabo entonces el estudio de fase III, KyberSept, estudio doble ciego, controlado y multicéntrico9. Un total de 2.314 pacientes con sepsis grave y shock séptico fueron asignados aleatoriamente a dos grupos iguales de 1.157 pacientes para recibir AT intravenosa (30.000 UI en 4 días) o placebo en las 6 horas del comienzo de la sepsis. La mortalidad a los 28 días fue similar en los dos grupos (38,9 frente al 38,7%; p = 0,94). En el subgrupo de pacientes que no recibieron heparina concomitante se observó un descenso significativo de la mortalidad a los 90 días (44,9% frente al 52,5%; p = 0,03). Este hallazgo es compatible con el efecto inhibidor de la heparina mencionado anteriormente y sugiere que en pacientes que no reciban heparina concomitante, la AT puede ser potencialmente útil en la sepsis grave, aunque sería necesario llevar a cabo un estudio dirigido a demostrar esta presunción.
INHIBIDOR DE LA VIA DEL FACTOR TISULAR EN LA SEPSIS GRAVE
En algunos estudios preclínicos se observó que la administración de TFPI recombinante (Tifacogin) mejoraba el pronóstico en la sepsis. Establecida la seguridad y bioactividad del fármaco en diversos estudios fase 2, se llevó a cabo un estudio fase 3 en pacientes con sepsis grave. El Tifacogin es producido en Escherichia oli y se diferencia del TFPI endógeno en una alanina y en la ausencia de glucosilación. El estudio OPTIMIST10, randomizado, doble ciego y multicéntrico, incluyó un total de 1.754 pacientes con sepsis grave e INR (international normalized ratio) mayor o igual a 1,2 y 201 pacientes con INR < 1,2. El objetivo de este grupo de bajo INR era evaluar la seguridad del tratamiento con Tifacogin. La mortalidad a los 28 días en los pacientes con INR elevado fue similar en los dos grupos (tifacogin 34,2% frente a placebo 33,9%, p = 0,88). Los pacientes que no recibieron tratamiento concomitante con heparina parecían beneficiarse más de la eficacia del Tifacogin, dato consecuente con el hecho de que el TFPI tiene dominios que ligan heparina que actúa desplazando el TFPI del endotelio. A la vista de estos hallazgos, sería deseable la realización de un estudio con Tifacogin en pacientes con sepsis grave y que no reciban heparina concomitante.
TRATAMIENTO DE LA SEPSIS CON PROTEÍNA C ACTIVADA
En la sepsis el endotelio ya no produce niveles adecuados de trombomodulina y del receptor de la PC endotelial por lo que se produce una deficiencia en la conversión a PCA. La PCA tiene un efecto antitrombótico ya que inactiva los factores de coagulación Va y VIIIa disminuyendo así la producción de trombina. La PCA al inhibir así la producción de trombina tiene un efecto antiinflamatorio indirecto, ya que la trombina estimula la activación plaquetaria, la adhesión del neutrófilo y la activación del endotelio. Otro efecto antiinflamatorio de la PCA depende de su acción en los monocitos que disminuyen entonces la producción de IL-1, del TNF-alfa, del factor inhibidor de la migración (MIF) y desacopla la interacción del LPS con el receptor CD-14 monocítico. La PCA disminuye la producción de la sintetasa inducible del óxido nítrico (iNOS). La PCA es profibrinolítica al inactivar el PAI-1 y el TAFI, ambos inhibidores endógenos de la fibrinólisis. Esta suma de efectos anticoagulante, antiinflamatorio y profibrinolítico serían beneficiosos para revertir la microtrombosis circulatoria de los pacientes con sepsis.
En 48 pacientes consecutivos con sepsis, Lorente et al7 observaron que todos los pacientes tenían niveles elevados de dímero D y complejos trombina-antitrombina, siendo los niveles de PC significativamente menores en los sépticos, fuesen infecciones por grampositivos o por gramnegativos, e igualmente en los supervivientes los niveles de PC empezaban a recuperarse desde el día 4 en adelante. La persistencia durante días de valores bajos de PC en los pacientes sépticos contrasta con las elevaciones transitorias de la mayoría de las citocinas, que además no tienen valor pronóstico consistente. En pacientes con shock séptico la presencia de coagulación intravascular diseminada (CID) se asociaba a una mayor mortalidad (77% frente al 32%) y a mayor número de órganos disfuncionantes desde el ingreso, y todos los pacientes excepto dos tenían niveles bajos de PC. Estos niveles bajos de PC gradualmente retornan a la normalidad en los pacientes que sobreviven a la sepsis. Pacientes traumáticos, neuroquirúrgicos o sépticos tienen alteraciones de la hemostasia aunque estas alteraciones son mucho más profundas en los sépticos que además tienen los niveles más bajos de proteína C. En el 90% de pacientes con sepsis grave se han observado niveles bajos de PC en el momento del diagnóstico, incluso en aquellos pacientes con tiempo de protrombina (TP), tiempo parcial de tromboplastina activada (TPTA), plaquetas y fibrinógeno normales, y esta disminución a los dos días se correlacionaba con la mortalidad a los 30 días. Estas observaciones sugieren que el consumo de PC se produce antes que se consuman otros factores como antitrombina, fibrinógeno y factor V. Este descenso en las cifras de PC se produce 12 horas antes de que el paciente desarrolle los signos de sepsis en pacientes neutropénicos y no está presente en aquellos que sólo desarrollan fiebre sin infección. Esta coagulopatía de consumo silente, previa a los signos francos de sepsis severa, se caracteriza por el descenso de los niveles de PC, sugiriendo la hipótesis de que el consumo de PC es un fenómeno patogénico en el desarrollo de la sepsis grave. Quizás el modelo más paradigmático de sepsis grave y alteraciones de la coagulación sea el shock séptico meningocócico con púrpura fulminante y en estos pacientes se ha demostrado una deficiencia adquirida de PC producida por la endotoxemia que daría lugar a un consumo masivo de PC. Disminuciones similares de PC se han observado en otras infecciones por gramnegativos como la fiebre tifoidea, la fiebre botonosa mediterránea, la meloidosis, la púrpura fulminante estreptocócica y en la malaria por Plasmodium falciparum, en la que además, una menor actividad de PC se correlacionaba con mayor parasitemia y mayores niveles de TNF-alfa.
La PCA administrada a babuinos tras una infusión de E. coli disminuía la mortalidad, y la administración de un anticuerpo a PCA la aumentaba. Resultados similares se observaron en ratas y en conejos tras la administración de endotoxina. También en babuinos, la coadministración de una dosis letal de E. coli y de anticuerpos al receptor endotelial de PC aumentaba la coagulación, la inflamación y la muerte, sugiriendo la importancia de un número adecuado del receptor endotelial a PC para la activación.
La drotrecogina alfa activada es una versión recombinante de la PCA plasmática humana producida en la línea celular HEK293 de riñón humano. Esta molécula difiere de la PCA endógena en algunos oligosacáridos y tiene propiedades similares. Tanto la PCA plasmática como la forma recombinante tienen una gran especificidad de especies y por tanto se requiere precaución para extrapolar las dosis de estudios animales al ser humano. No se han identificado efectos secundarios, salvo los relacionados con su actividad antitrombótica, en los estudios de toxicología animal, incluyendo prolongación de los tiempos de protrombina y de tromboplastina activada. Los estudios clínicos llevados a cabo indican que la drotrecogina alfa activada tiene un pequeño volumen de distribución con una eliminación bifásica que alcanza una concentración plasmática constante a las dos horas de iniciar una infusión intravenosa continua y el 90% se elimina a las dos horas de interrumpir la infusión. El aclaramiento plasmático depende del peso corporal y es apropiado el ajuste de dosis basado en el peso. No se requieren ajustes de la dosis dependiendo del sexo, edad, función renal, hepática o niveles endógenos de PC.
En los estudios de fase II realizados en pacientes con sepsis grave no se observaron diferencias en la incidencia de efectos adversos y hemorragia entre el grupo placebo y el grupo tratado con drotrecogina alfa activada. Se demostró una relación significativa entre la dosis y la disminución de dímero D y concentración de IL-6, eligiéndose la dosis de 24 µg/kg/ h durante 96 horas para la realización del estudio de fase III11. Esta dosis producía concentraciones plasmáticas suprafisiológicas de PCA y es, por tanto, una intervención farmacológica y no una terapia de reemplazamiento.
El estudio de fase III (PROWESS)12 se programó para evaluar la mortalidad por cualquier causa a los 28 días con un reclutamiento total de 2.280 pacientes con sepsis grave. Los pacientes se randomizaron para recibir drotrecogina alfa activada (24 µg/kg/h) o placebo durante 96 horas. Para la inclusión en el estudio, los pacientes no podían tener una disfunción orgánica relacionada con la sepsis durante más de 24 horas y el tratamiento debía comenzar en las 24 horas siguientes a la randomización. Un comité de evaluación externo sugirió poco después de empezar el estudio que un gran número de los pacientes incluidos en el estudio tenían un elevado riesgo de muerte por causas no relacionadas con la sepsis: neoplasias terminales, encefalopatía postanóxica, enfermedades subyacentes, etc., por lo que se aprobó por la Food and Drug Administration (FDA) una enmienda para disminuir el número de pacientes con estas patologías. Los análisis interinos llevados a cabo por un consejo independiente recomendaron la interrupción del estudio por eficacia cuando se habían evaluado 1.520 pacientes.
Los pacientes tratados con drotrecogina alfa activada experimentaron una reducción significativa de la mortalidad a los 28 días (24,7% frente al 30,8%; p = 0,005), esto es, una reducción absoluta del 6,1% en el número de muertes y una reducción del 19,4% en el riesgo relativo de muerte. Más del 85% de los pacientes reclutados en el estudio tenían bajas concentraciones plasmáticas de PC y, en estos pacientes, la disminución del riesgo de muerte era del 20%. Poco más del 10% de los pacientes tenía concentraciones normales de PC y en este grupo la disminución del riesgo de muerte era del 42%. Igualmente se observó un acortamiento en el período de reversión del shock y de la disfunción respiratoria, aumentando el número de días con vida y libres de shock o ventilación mecánica.
La actividad antitrombótica de la drotrecogina alfa activada quedó de manifiesto por la disminución que se observó en los valores de dímero D durante la primera semana desde el comienzo del tratamiento. Los niveles de PAI-1 también disminuyeron más en el grupo tratado con drotrecogina alfa hacia el quinto día, aumentando simultáneamente los niveles de plasminógeno y restaurando la actividad fibrinolítica normal. Los niveles de IL-6, marcador de actividad inflamatoria en la sepsis, también bajaron más en el grupo tratado. A la vista de estos resultados quedó demostrada clínicamente la eficacia de la drotrecogina alfa activada como agente antitrombótico, fibrinolítico y antiinflamatorio.
La reducción absoluta de la mortalidad fue del 13% en los pacientes con un APACHE > 24, más del doble de la disminución global de la mortalidad en la población total del estudio. La FDA aprobó el uso de la drotrecogina alfa activada en pacientes con sepsis grave y un APACHE > 24 y ha dispuesto la realización de un estudio en pacientes con sepsis y una sola disfunción orgánica que se está realizando actualmente. En otros países como España, el medicamento Xigris®, nombre comercial de la drotrecogina alfa activada, se ha aprobado para su uso en pacientes con sepsis grave y disfunción de al menos dos órganos.
Los estudios clínicos llevados a cabo con drotrecogina alfa han mostrado más muertes asociadas a hemorragia en los pacientes tratados con drotrecogina alfa en comparación con los que recibieron placebo (6 frente a 2). La mayoría tenían trombocitopenia intensa (menos de 30.000/µl). La incidencia de hemorragia es mayor en los pacientes tratados sólo durante el período de infusión del fármaco (3,6% frente al 2,0%), siendo similar después de la infusión. Esta mayor incidencia de sangrado se asoció exclusivamente a procedimientos invasivos, siendo la incidencia de hemorragias espontáneas similar en los dos grupos. Ante este riesgo aumentado de sangrado, las recomendaciones de uso de la drotrecogina alfa activada aconsejan valorar los riesgos frente a los potenciales beneficios en los pacientes que estén recibiendo simultáneamente heparina (> 15 UI/kg/h) o hayan recibido recientemente trombolíticos (últimos 3 días), anticoagulantes orales/inhibidores de glucoproteína IIb/IIIa/ácido acetilsalicílico (> 650 mg)/antiagregantes plaquetarios (últimos 7 días) así como en los pacientes con riesgo aumentado de sangrado tales como hemorragia digestiva reciente (últimas 6 semanas), ictus isquémico (últimos 3 meses), malformación arteriovenosa o aneurisma cerebral, diátesis hemorrágica no relacionada con la sepsis, hepatopatía crónica grave o pacientes con sangrado activo y no controlado. Se recomienda interrumpir la infusión 2 horas antes de procedimientos invasivos, reiniciándola 12 horas después de cirugía mayor o inmediatamente si son procedimientos menores.
La infusión de drotrecogina alfa tiene una mínima influencia sobre el tiempo de protrombina, y su efecto es variable sobre el tiempo de tromboplastina activada, por lo que no se debe emplear éste para valorar su efecto farmacodinámico. Menos del 1% de los pacientes con sepsis grave que reciben drotrecogina alfa activada forman anticuerpos anti-PCA. Estos anticuerpos no neutralizan el efecto de la PCA en el tiempo de tromboplastina activada. En pacientes sanos no se ha observado formación de anticuerpos incluso después de 6 cursos de tratamiento12.
Uno de los aspectos a considerar en la implementación de nuevas terapias es el coste-beneficio de su aplicación. Con un coste cercano a los 7.000 euros por un tratamiento completo de 96 horas o 4 días en un paciente de unos 70 kg, resulta obligado preguntarse por la justificación del mismo. En un medio en el que el gasto sanitario sube muy por encima de la inflación existe una presión razonable para justificar económicamente los tratamientos administrados y valorarlos teniendo en cuenta su eficacia. En cuanto a la eficacia podemos compararla con otros estudios de terapias ampliamente aceptadas en nuestra práctica médica. El estudio GISSI, comparando estreptokinasa con placebo en el infarto agudo de miocardio (IAM), demostró una disminución absoluta de la mortalidad del 2,3%, lo que supone tratar 43 pacientes para salvar una vida. El ISIS-2, comparando estreptokinasa más ácido acetilsalicílico frente a placebo en el IAM demostró una reducción de la mortalidad del 5,2%, implicando el tratamiento de 19 pacientes para salvar una vida. El GUSTO, comparando activador tisular del plasminógeno frente a estreptokinasa, encontró una disminución de la mortalidad del 1%, lo que supone tratar 100 pacientes para salvar una vida. Estas disminuciones de la mortalidad están por debajo de la observada en el PROWESS que fue del 6,1%, por tanto, se necesita tratar sólo 16 pacientes para salvar una vida. En pacientes con disfunción de dos órganos o una población con un APACHE mayor de 24, la reducción de la mortalidad es del 13%, esto es, se salva una vida por cada 8 pacientes tratados.
El coste calculado en EE.UU. de la drotrecogina alfa activada en pacientes con sepsis grave para la indicación aprobada por la FDA (APACHE > 24) es de 27.000 $ (año ganado ajustado a la calidad de vida), sensiblemente inferior a terapias actuales utilizadas en la práctica médica como el desfibrilador implantable (40.000 $), estatinas para la prevención primaria y secundaria (48.000 $), los cuidados neonatales para niños con peso menor de 1 kilo (49.000 $), el by pass aortocoronario para enfermedad de un solo vaso (56.000 $), el trasplante de pulmón (200.000 $) o la resucitación cardiopulmonar intrahospitalaria (250.000 $) y extrahospitalaria (> 600.000 $)13.
Por tanto desde el punto de vista de la eficacia y del coste-beneficio, la drotrecogina alfa activada es un tratamiento recomendado para pacientes con sepsis grave que presentan disfunción de al menos dos órganos.

