





Editado por: F. Baigorri-González y J.º. Lorente Balanza
Última actualización: Abril 2005
Más datosINTRODUCCION
La sepsis continúa siendo una de las principales causas de muerte en los pacientes críticos y su incidencia ha mostrado un lento pero sostenido crecimiento en los últimos años. De manera frecuente se asocia con fracaso orgánico múltiple que puede ser grave y progresivo determinando, en un alto porcentaje de casos, el fallecimiento del paciente1-3. El aumento de la incidencia del shock séptico puede atribuirse a varios factores: mayor número de pacientes inmunodeprimidos, aumento de procedimientos diagnósticos invasivos, prolongación de la expectativa de vida y mayor resistencia de los microorganismos a los antibióticos. Adicionalmente, algunos pacientes contraen esta entidad dentro de las Unidades de Cuidados Intensivos, como complicación asociada a diferentes medidas y procedimientos invasivos4.
Los mecanismos involucrados en el desarrollo del daño orgánico son multifactoriales y todavía no bien conocidos. En algunas circunstancias, la disfunción orgánica es atribuible a la hipoxia tisular como consecuencia del deterioro hemodinámico y disminución de la perfusión o isquemia tisular. Sin embargo, el papel de la hipoxia tisular ha sido cuestionado como mecanismo exclusivo del fallo orgánico en la sepsis5. Así, en ausencia de compromiso hemodinámico demostrable, como puede ocurrir en los estados hiperdinámicos del shock séptico, resulta cuestionable atribuir el fallo orgánico a hipoxia tisular. Sin embargo, aún es posible que puedan ocurrir trastornos de la distribución del flujo sanguíneo y alteraciones graves en la microcirculación que comprometan la perfusión tisular efectiva6,7.
Por otra parte, hay evidencia de otros posibles mecanismos patogénicos. La activación de leucocitos polimorfonucleares, característica del proceso inflamatorio que acompaña a la sepsis, cursa con un marcado incremento del estrés oxidativo, entendiendo por tal el disbalance entre la formación de especies reactivas del oxígeno (ERO) y las defensas antioxidantes, en favor de las primeras8. Las ERO no neutralizadas por las defensas antioxidantes reaccionan con proteínas, ADN, ARN y lípidos de membranas celulares determinando bloqueos en pasos del metabolismo celular y disfunción mitocondrial, que conducen al fracaso energético celular9.
Simultáneamente con el proceso inflamatorio ocurre aumento de la producción de óxido nítrico (NO) por inducción de la óxido nítrico sintetasa inducible (iNOS). La superproducción de NO determina su reacción, mediada por difusión, con el anión superóxido dando peroxinitrito (ONOO-), especie altamente reactiva capaz de oxidar y nitrar componentes celulares y tisulares, tales como los residuos de tirosina de las proteínas celulares y plasmáticas, el ADN y lípidos o enzimas críticas del metabolismo intermediario10. Así, está bien documentado el daño estructural y funcional a nivel mitocondrial, que se torna irreversible debido a oxidación y nitración de componentes mitocondriales. El ONOO- también oxida y produce depleción de antioxidantes endógenos tales como ascorbato, glutatión y superóxido dismutasa10. La presencia de ONOO- y otras especies reactivas derivadas del nitrógeno (ERN) tales como el dióxido de nitrógeno (NO2), cuyo papel en la sepsis es objeto de investigación reciente, nos lleva a ampliar el concepto clásico de estrés oxidativo al de estrés nitrosativo. En esta situación, predomina la formación de estas ERN sobre los mecanismos detoxificadores intracelulares y plasmáticos, llevando al consiguiente daño celular y tisular. El estrés oxidativo y nitrosativo actúan perpetuando el proceso inflamatorio por diferentes mecanismos. Las ERO y ERN tienen efectos quimiotácticos, favoreciendo el reclutamiento de neutrófilos. Además, las moléculas mediadoras de estrés oxidativo se constituyen también en mensajeros intracelulares para la transducción de señales del proceso inflamatorio. Así por ejemplo, el factor nuclear kappa B (NFκB) activado por estos mensajeros secundarios migra al núcleo donde selectivamente estimula la transcripción de proteínas específicas de la inflamación. Éste induce la transcripción de genes que promueven la producción de citocinas como la interleucina-6 (IL-6), la interleucina-8 (IL-8) y moléculas de adhesión (ICAM-1) que agudizarán o perpetuarán el proceso inflamatorio11.
En la búsqueda de nuevas medidas terapéuticas para el manejo de la sepsis grave y del shock séptico se han producido algunos avances. La administración oportuna de soporte nutricional, la prevención de las infecciones nosocomiales, la prevención de úlceras de estrés, la profilaxis de la trombosis venosa profunda, el uso adecuado de sedación-analgesia y el uso de patrones de ventilación mecánica protectora, han desempeñado un papel importante y podrían ser beneficiosos en algunos subgrupos de pacientes4. Tratamientos farmacológicos más específicos, que intentaron modular o bloquear algunos componentes del proceso inflamatorio, no alcanzaron el éxito esperado o fracasaron decididamente. Sin embargo, los conocimientos sobre la bioquímica de la sepsis han aumentado de manera sustancial en los últimos años, abriendo expectativas al ensayo de nuevas terapias alternativas. Siendo el estrés oxidativo y nitrosativo un capítulo fundamental en la patogenia de la sepsis, la terapia moduladora de la síntesis de NO, el empleo de antioxidantes y/o de atrapadores de ERO y ERN, han mostrado beneficios prometedores en modelos experimentales. No es aventurado pensar entonces que un enfoque de este tipo resulte finalmente en una terapia exitosa para disminuir la mortalidad de pacientes con shock séptico.
ESPECIES REACTIVAS DERIVADAS DEL OXIGENO Y DEL NITRÓGENO
El oxígeno es esencial para la vida de los organismos aerobios, y su mayor parte (98% o más) es utilizado para la generación intracelular de energía, proceso en el cual resulta reducido completamente a agua, involucrando la transferencia de 4 electrones a nivel de la cadena respiratoria mitocondrial (fig. 1). Sin embargo, entre los nutrientes iniciales y la generación de energía, se forman varias moléculas por reducción parcial del oxígeno, tales como el radical anion superóxido (O2-) y el peróxido de hidrógeno (H2O2). La reacción entre estas dos especies en presencia de metales (hierro intracelular), lleva a la formación de otro radical, el radical hidroxilo (OH), uno de los oxidantes más potentes que existen.
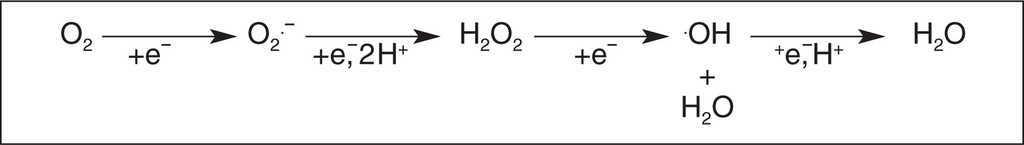
Figura 1. Reacciones de formación de especies reactivas del oxígeno (ERO).
Debido a su corta vida media, baja selectividad y elevada reactividad, los blancos biológicos de las ERO incluyen diversos componentes tanto intra como extracelulares tales como proteínas, enzimas, ácidos nucleicos, membranas y lipoproteínas.
Fuentes intracelulares
Las fuentes intracelulares de ERO incluyen los complejos I y II de la cadena mitocondrial de transporte electrónico, peroxisomas, sistemas enzimáticos de mono y dioxigenasas y los leucocitos polimorfonucleares. En los últimos años se ha demostrado que ante diferentes estímulos fisiopatológicos, las células vasculares producen especies reactivas del oxígeno y que la principal fuente de radicales libres en estas células es una oxidasa vascular que utiliza NADH y NADPH como sustratos reductores para la transferencia monoelectrónica al oxígeno molecular y producción concomitante de O2-. Esta oxidasa vascular es, en muchos aspectos, similar a la NADPH oxidasa de los neutrófilos, aunque difiere en sus mecanismos de activación. Un aspecto particularmente importante de la NADH/NADPH oxidasa vascular es que su actividad es estimulada por angiotensina II (a través del receptor AT1), citocinas (por ejemplo factor de necrosis tumoral alfa [TNF-α]) y lipoproteínas oxidadas. A través de la estimulación de dicha oxidasa vascular, la angiotensina II se constituye en una causa principal de estrés oxidativo vascular, con la consiguiente generación de disfunción endotelial, vinculado estrechamente a las etapas iniciales del proceso séptico.
Defensas antioxidantes
Los organismos aerobios están expuestos a un estado estacionario basal de oxidantes y las células se hallan protegidas contra esta producción basal por una serie de sistemas antioxidantes de naturaleza enzimática (superóxido dismutasa, catalasa, peroxidasas) y no enzimática. La mayoría de los antioxidantes enzimáticos se basan en principios de reacciones redox y están localizados en medios acuosos (vitamina C, glutatión) o lipídicos (vitamina E, ubiquinol, carotenoides y flavonoides). El α-tocoferol, componente principal de la vitamina E, constituye el principal antioxidante liposoluble capaz de interrumpir la oxidación de lípidos insaturados a nivel de membranas biológicas y lipoproteínas, preservando su integridad estructural y funcional. Cuando un radical lipídico del tipo alcoxilo (LO) o peroxilo (LOO) generado por procesos de lipoperoxidación reacciona con α-tocoferol, es reducido a hidroperóxido lipídico (LOOH), incapaz de seguir reacciones de propagación de la lipoperoxidación (fig. 2). De esta forma el α-tocoferol al donar un electrón y oxidarse como consecuencia de neutralizar radicales lipídicos, se transforma en radical tocoferoxilo, que puede ser reconvertido a su estado nativo tocoferol por otros antioxidantes. La vitamina C o ácido ascórbico (ascorbato) es el reductor más importante de α-tocoferol, con el coste de la formación de un radical de la vitamina C (ascorbilo) que, a su vez, puede ser reducido nuevamente a ascorbato por otros reductores como glutatión y tioles. La acción combinada de las vitaminas E y C resulta en un sinergismo antioxidante debido a su localización en distintos compartimientos (fase liposoluble e hidrosoluble, respectivamente). Debido a sus propiedades reductoras, el ascorbato neutraliza distintas ERO y ERN. En el curso de estas reacciones, el ascorbato se oxida a dihidroascorbato, siendo capaz de ser reciclado nuevamente a ascorbato por reductores tales como el glutatión. Por último, tenemos oligoelementos que pertenecen al grupo de defensas antioxidantes no enzimáticas, participando como cofactores esenciales para la acción de enzimas antioxidantes: el selenio es cofactor de la enzima detoxificadora de peróxido de hidrógeno glutatión peroxidasa, y el zinc es cofactor de la superóxido dismutasa citosólica (Cu-Zn SOD), responsable de la eliminación de radicales superóxido.
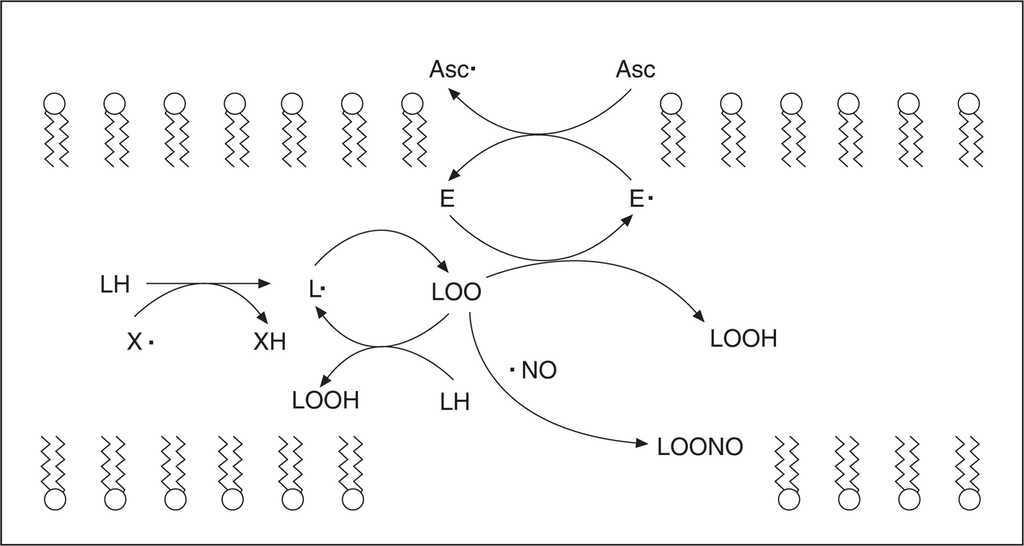
Figura 2. Mecanismo de lipoperoxidación y acción antioxidante de las vitaminas C, E y del óxido nítrico.
Oxido nítrico
Por otra parte, estudios de nuestro laboratorio12,13 han permitido postular al NO como principal antioxidante lipofílico endógeno, mediante su capacidad de difundir a medios hidrofóbicos y terminar reacciones de propagación de la lipoperoxidación, actuando con el α-tocoferol para la eliminación de los radicales lipídicos, preservando a la vitamina E del daño oxidativo (fig. 3). El NO constituye el principal componente químico del factor de relajación dependiente del endotelio (EDRF) que produce la relajación del músculo liso, siendo sintetizado en forma constitutiva por la óxido nítrico sintetasa endotelial (eNOS) a partir del aminoácido L-arginina, existiendo además la forma constitutiva neuronal (nNOS) y la forma inducible (iNOS). Desde el punto de vista cardiovascular el efecto vasodilatador es la función más importante del NO, pero también se sabe que como mensajero es una molécula de múltiples funciones reguladoras en diferentes sistemas. Por su estructura, el NO es un radical libre centrado en nitrógeno y como tal puede participar en reacciones con otras especies reactivas del oxígeno (fig. 3).
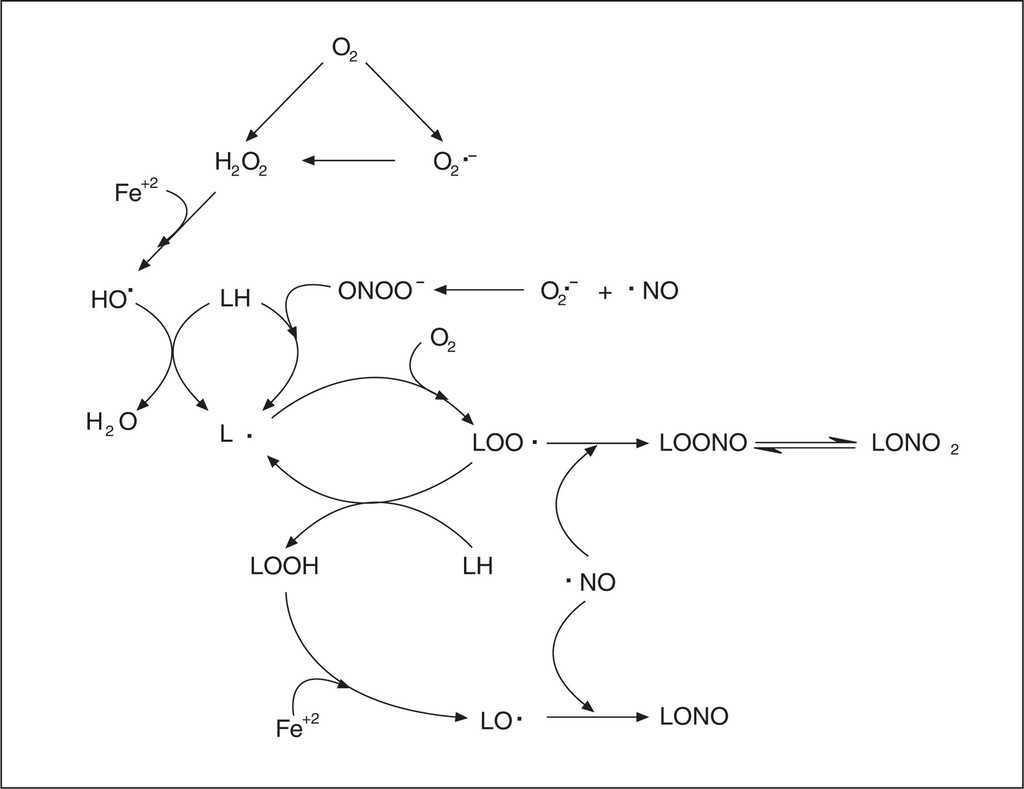
Figura 3. Reacciones pro y antioxidantes del oxído nítrico en membranas.
De particular importancia para la biología vascular es la reacción limitada por difusión entre el O2- y el NO (KM 1010 M-1s-1)10. Esta reacción produce un agente fuertemente oxidante y nitrante, el peroxinitrito (ONOO-). La reacción radical-radical entre el O2- y el NO es más rápida que la reacción entre el O2- y la superóxido dismutasa (k = 1,9 x 109 M-1s-1). El interjuego entre la producción de O2-, su eliminación por la superóxido dismutasa y su reacción con el NO, posee un papel central en la modulación de la actividad biológica del NO. El ONOO- en su forma aniónica es capaz de protonarse a pH fisiológico para dar ácido peroxinitroso (ONOOH) que rinde OH y NO2, o se descompone a nitrato (NO3-)10. Es así que el ONOOH inicia procesos de lipoperoxidación al abstraer un electrón de un ácido graso insaturado (LH)12. El producto de la reacción es un radical lipídico (L), que a su vez en presencia de oxígeno molecular es capaz de generar reacciones de propagación en cadena, formando radicales peroxilo de ácidos grasos (LOO). En estas situaciones el NO está desempeñando un papel prooxidante debido a que el ONOO- generado es un agente fuertemente oxidante y nitrante a nivel de proteínas y lípidos de membrana e intracelulares10,12. La faz antioxidante del NO ocurre cuando interviene en reacciones radical-radical de terminación de los procesos de lipoperoxidación limitadas por difusión (LOO + NO, KM 109 M-1s-1), dando productos nitrogenados no radicales10,12,14. El comportamiento del NO como pro o antioxidante depende de la concentración de O2- que, cuando es elevada, aumenta las reacciones oxidativas. La corta vida media del NO y el delicado balance existente entre su síntesis, utilización por blancos biológicos normales o por el contrario, por reacciones que median su descomposición, inactivación o transformación secundaria en ERN, determina que haya situaciones en las que su disponibilidad esté disminuida, favoreciendo y generando procesos de daño tisular. Estas situaciones pueden determinar una síntesis disminuida y/o un consumo aumentado. La síntesis de NO puede estar disminuida por una alteración en la concentración de sustratos para la NOS en la célula endotelial, presencia de inhibidores endógenos de esta enzima (ADMA), así como por alteración de la expresión de la enzima, que si bien es constitutiva, está sujeta a regulación por diferentes mecanismos. La generación aumentada de ERO desvía al NO de su metabolismo normal y no sólo altera su disponibilidad, sino que lleva a la producción de ERN tales como el ONOO-.
HIPOXIA CITOPÁTICA Y DISFUNCIÓN MITOCONDRIAL
Varios investigadores han señalado la existencia de trastornos del metabolismo energético celular en la sepsis. El mismo estaría en la base de la disfunción y muerte celular y, por tanto, sería responsable también de la disfunción orgánica múltiple. El término hipoxia citopática ha sido acuñado para referirse a esta situación donde el daño o el bloqueo de pasos metabólicos intracelulares desempeña un papel fundamental9.
La disminución o bloqueo de la respiración mitocondrial por inactivación de distintos complejos de la cadena respiratoria o la depleción de depósitos celulares de NAD+/NADH por activación de la enzima nuclear Poly-ADP-ribosa polimerasa son mecanismos posibles de la falla energética celular15.
Hemos mencionado el hecho de que las mitocondrias son el blanco preferido de la hiperproducción de ERO y ERN. El daño en la respiración mitocondrial ha sido demostrado en diversos modelos experimentales y también en seres humanos. Esta agresión mitocondrial tiene manifestaciones funcionales y estructurales evidentes, habiéndose encontrado una buena correlación anatomofuncional16-18.
En seres humanos, Brealey et al mostraron la asociación entre disfunción mitocondrial e intensidad del shock séptico17. Estos autores estudiaron biopsias de músculo esquelético en un grupo de pacientes sépticos, midiendo la respiración mitocondrial en las muestras recogidas. Los resultados se compararon contra biopsias de una población de sujetos no sépticos, encontrándose disminución de la actividad de la cadena respiratoria, descenso de concentraciones intracelulares de adenosina trifosfato (ATP), descenso de glutatión intracelular y aumento de concentración de nitritos/nitratos (productos finales de la descomposición del NO en los sujetos sépticos). Hubo asociación entre sobreproducción de NO, depleción de antioxidantes, disfunción mitocondrial y descenso de concentraciones de ATP, con el nivel de gravedad del fallo orgánico y con la mortalidad de los casos. Boulos et al también mostraron depresión de la respiración mitocondrial cuando estos organelos eran expuestos a suero de pacientes sépticos. Ello podía ser atenuado por inhibición de la iNOS y de la sintetasa Poly-ADP-ribosa18.
En un modelo experimental de sepsis peritoneal hemos demostrado depresión de la respiración mitocondrial en diafragma, que se asocia a disminución de antioxidantes en plasma y aumento en la producción de NO, lipoperoxidación y nitración de proteínas plasmáticas y mitocondriales19. Esta disfunción mitocondrial se asocia con un descenso de las fuerzas diafragmáticas estudiadas in vitro19, enfatizando que la hiperproducción de NO y el consiguiente incremento de actividad de ERN desempeñan un papel protagonista en la génesis de la disfunción multiorgánica.
Las mitocondrias constituyen un blanco intracelular para la acción del NO, dada la fácil difusión de éste a través de las biomembranas. El NO puede alcanzar las mitocondrias difundiendo de fuentes extramitocondriales de la misma célula o desde células adyacentes. Puede actuar de forma autocrina o paracrina e interactuar con los componentes mitocondriales. Niveles fisiológicos de NO (10-100 nM) pueden producir una completa inhibición de la respiración mitocondrial a concentraciones normales de O2 tisular16. Una de las causas de este fenómeno es la interacción del NO con la citocromo oxidasa, reduciendo los componentes de la cadena de transporte de electrones. A su vez, el NO inhibe los complejos I y II16. En el caso del complejo I, el NO por sí mismo es capaz de inhibir este complejo en la mitocondria aislada. La inhibición mediada por el NO requiere la formación transitoria de nitrosotioles y ONOO-, que pueden nitrar u oxidar grupos tioles del complejo I. En el caso del complejo II, el NO puede unirse en los cluster hierro-sulfuros, pero sólo son observados efectos significativos ante grandes concentraciones de NO16. A su vez el NO puede reaccionar con el ubiquinol para generar NO- y radical ubisemiquinona20. Esta reacción favorece la producción de ONOO-, por lo que contribuiría al daño oxidativo mitocondrial. Todos estos mecanismos de daño mitocondrial se podrían desencadenar y/o sobreproducir en la sepsis, determinando la disminución de la actividad de los complejos de la cadena respiratoria, con el consiguiente bloqueo en la producción de ATP.
TERAPIA ANTIOXIDANTE EN LA SEPSIS
El estrés oxidativo y nitrosativo y su consecuencia, la hipoxia citopática, podría explicar el fracaso de distintas estrategias empleadas en el manejo clínico del shock séptico. Así, puede explicarse que los esfuerzos dirigidos a mejorar la oxigenación tisular por aumento del aporte sistémico de oxígeno o la optimización de la función cardiovascular resulten infructuosos cuando la maquinaria mitocondrial está bloqueada.
La terapia antioxidante en el tratamiento de la sepsis o de otras entidades del enfermo crítico como el daño por isquemia-reperfusión tiene todavía importantes desafíos por delante. En primer lugar, es posible actuar a diferentes niveles como puede ser la síntesis de NO y la producción de ERO o ERN, o tratando de amortiguar los efectos celulares o tisulares de esta sobreproducción. Hay evidencia experimental de que los tratamientos antioxidantes pueden generar beneficio en la sepsis, mejorando la respiración mitocondrial y la disfunción orgánica21.
Las estrategias antioxidantes pueden ser variadas. Se ha ensayado aumentar el aporte exógeno de vitaminas como el α-tocoferol, el ß-caroteno o el ácido ascórbico21,22. Otros compuestos con acción antioxidante como la N-acetilcisteína han mostrado resultados alentadores11. Oligoelementos tales como el selenio, cobre, zinc y manganeso también han sido investigados, pero no hay resultados definitivos sobre posibles beneficios en pacientes críticos.
Recientemente se ha propuesto el uso de una nueva generación de compuestos antioxidantes con acción superóxido dismutasa símil, las metaloporfirinas, que actúan catalíticamente como atrapadores de O2-, ONOO-, NO2, LOO y otras especies reactivas23. Se ha demostrado su acción antioxidante tanto in vitro como in vivo10,24. En nuestro modelo experimental, hemos podido demostrar mejoría tanto de la disfunción muscular diafragmática como de la respiración mitocondrial con el empleo profiláctico de Mn(III) porfirinas19. Con un enfoque conceptual similar, se han empleado estas sustancias en la disfunción renal de la sepsis con resultados alentadores sobre la preservación del flujo sanguíneo y de la función renal25.
Si bien estos datos deben ser confirmados en series mayores, se abren grandes expectativas en el tratamiento de la disfunción orgánica de la sepsis a través de la modulación de la hiperproducción o la amortiguación de los efectos de las ERO y ERN26. Quedan preguntas por resolver sobre cuál es el mejor fármaco o la mejor combinación de fármacos a usar (por ejemplo metaloporfirinas + inhibidores de la iNOS), dosis más adecuada, vías de administración y, sobre todo, oportunidad del tratamiento. En este sentido, el tratamiento profiláctico, previo al desarrollo de la lesión oxidativa parecería ser la mejor estrategia. Se requiere mayor investigación sobre todos estos puntos con la certeza de que en el futuro se podrá disponer de agentes farmacológicos que ayuden a prevenir la disfunción multiorgánica (DMO) mediante el control del estrés oxidativo y nitrativo.
AGRADECIMIENTOS
Homero Rubbo es financiado por grants de Fogarty-NIH, Wellcome Trust (UK) y Guggenheim Memorial Foundation, así como Comisión Sectorial de investigación Científica de la Universidad de la República, Fundación Manuel Pérez-Facultad de Medicina y Programa de Desarrollo de las Ciencias Básicas, Uruguay.

