




Thrombotic microangiopathy (TMA) is a pathologic process characterized by inflammation and thickening of wall vessels, endothelial edema, basement membrane detachment, intimal fibrosis and platelet thrombi formation with occlusion of vascular lumen. It affects mainly kidney and central nervous system microvasculature1; depending on the lesion distribution, two different syndromes are described: Thrombotic Thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Both are characterized by microangiopathic hemolytic anemia and thrombocytopenia.2,3 Neurological disorders are dominant in TTP whereas renal failure is dominant in HUS.
HUS is denominated typical (90% of the cases) when caused by enteric shiga toxin-producing microorganisms, as Escherichia coli serotype O157:H7. The other rare cases are classified as atypical (aHUS), due to alternate complement pathway deregulation, or secondary to conditions such as pregnancy, autoimmune disease, invasive pneumococcal infections, HIV and H1N1 infections, cancer, malignant hypertension, and certain drugs.1,2
Most aHUS cases have a strong genetic component involving mutations and polymorphisms in the complement proteins genes.4 It is more common in children and young adults and the prognosis is poor with high morbidity and mortality in acute phase and 50% of progression to chronic kidney disease.1
The authors describe the case of a 35-year-old man who presented at the emergency department with fever, abdominal pain, nausea, bilious vomiting and decrease urine output for two weeks. He denied diarrhea and revealed history of intravenous drug abuse, under methadone program.
At admission he was pale, with icteric sclera, hypertensive (197/108mmHg) and tachypneic. Biochemistry revealed: normocytic normochromic anemia (Hb 6g/dL); platelet count 132×109L−1, acute kidney injury (Urea 81mmol/L; Creatinine 1487μmol/L); high Lactate dehydrogenase (LDH 23μkat/L) and proteinuria (100mg/dL). In the following hours the patient developed agitation, hallucinations and respiratory failure due to aspiration of gastric content, for what he was intubated and mechanically ventilated.
At the Intensive Care Unit he presented thrombocytopenia (platelets 98×109), severe kidney impairment with anuria and started hemodialysis. The anemia study revealed hemolysis: schistocytes in peripheral blood smear, haptoglobin <7mg/dL and raised LDH (26.7μKat/L), and the patient underwent regular blood transfusion, during the first week. TMA diagnosis was admitted and daily plasmaferesis was initiated, with exchange of 1.5 plasma volume per session. He completed a total of eleven sessions, with renal function recovery, allowing the spacing of dialysis sessions, as well as normalization of platelets count and LDH levels.
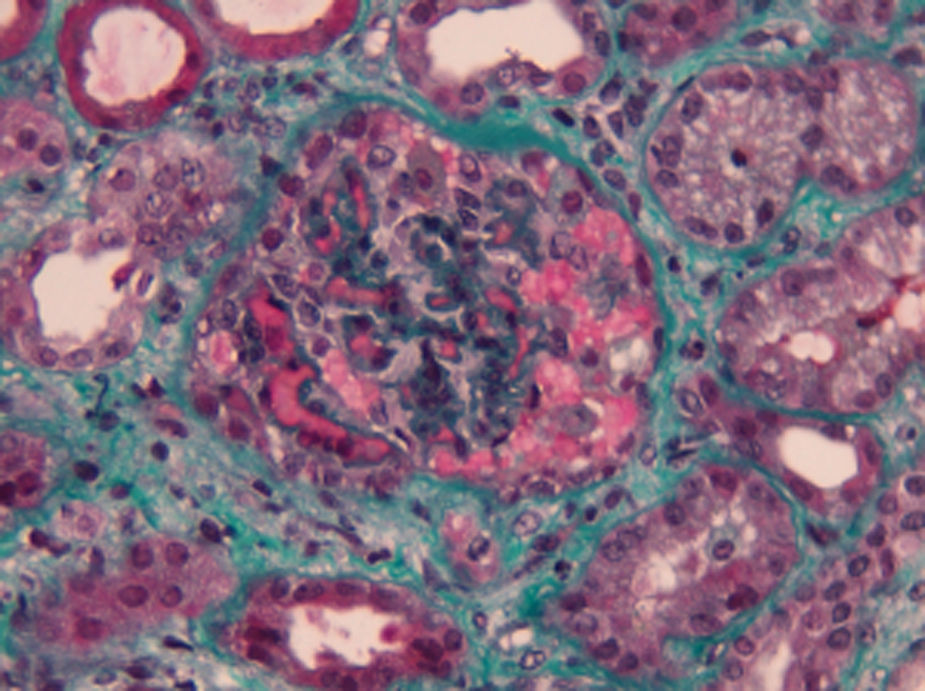
Complementary laboratory study excluded malignant hypertension, autoimmunity diseases, HIV and H1N1 infection. Shiga toxin was negative, C3 factor decreased, with no other changes in complement factors or respective antibodies. The activity of ADAMTS13 was normal without anti-ADAMTS13 antibodies (Table 1). The total body CT-scan excluded neoplasia. Kidney biopsy showed histopathological features of thrombotic microangiopathy (Fig. 1) and the genetic screening revealed a mutation in factor H.
Complementary laboratory study.
| Blood, urine and bronchial cultures | Negative |
| Shiga toxin | Negative |
| HIV antibodies | Negative |
| H1N1 (PCR) | Negative |
| Streptococos pneumoniae and Legionella pneumophila urine antigen | Negative |
| Antistreptolysin-O (ASO) titers | Negative |
| Complement study | |
| C3 factor | 0.48g/L [0.9–1.8] |
| C4, CHF, CHB, CHI | Normal |
| Anti-ADAMTS13 antibodies | Negative |
| ADAMTS13 activity | 73% (normal >6%) |
| ANA, anti-dsDNA, anti-Sm, anti-SSA (Ro), anti-SSB (La), anti-RNP, anti-cardiolipine, anti-beta-2 glycoprotein I, anti-phospholipid antibodies | Negative |
| Serum protein electrophoresis | Normal |
HIV: Human immunodeficiency virus; PCR: polymerase chain reaction; CFH: factor H; CFI: factor I; CFB: factor B; ADAMTS13: a desintegrin and metalloprotease with thrombospondin type1 domain 13; ANA: antinuclear antibody; dsDNA: double-stranded DNA; Sm: Smith; SSA: Sjögren syndrome A; SSB: Sjögren syndrome B; RNP: ribonucleic protein.
During the hospitalization the patient presented high blood pressure, requiring labetalol perfusion followed by various antihypertensive agents. On the seventh day, he was successfully extubated. As infectious complications, he had Staphylococcus epidermidis bacteremia and Stenotrophomonas maltophilia on sputum smear, both resolved with the appropriate antibiotherapy.
At the time of discharge, the platelet count was 275×109/L, LDH 4.48μkat/L, Urea 27.8mmol/L, Creatinine 223.6μmol/L. Due to the persistence of kidney dysfunction the patient was transferred to nephrology ward where he continued on hemodialysis and started treatment with eculizumab, in the dosis of 900mg weekly in the first month, followed by 1200mg every 2 weeks thereafter.
Hemolytic anemia, thrombocytopenia and acute renal failure as initial presentation were suggestive of TMA most likely HUS due to the magnitude of renal impairment. Subsequent finding of a regular ADAMTS13 activity excluded TTP.5 Absence of diarrhea on presentation, negative Shiga toxin made less probable typical HUS diagnosis. Moreover, drugs and pathological conditions that cause secondary HUS were excluded.
Atypical HUS associated with genetic or immune complement abnormalities was the most probable hypothesis. Activation of the alternative complement pathway with low serum C3, normal C4 and Factor B as well as absence of anti-factor H antibodies and normal levels of Factor H and I indicated a possible mutation in genes encoding C3, factor H or factor I,6 that was confirmed by the genetic screening.
Mutation in factor H was first identified in 1973 and it courses with low or normal plasma levels of factor H.6 It is the most frequent genetic abnormality in aHUS patients (20–30%) and is associated with 50–70% risk of death or end stage renal disease in the first episode (<1 year) with a 50% risk of relapse and 75–90% risk of recurrence after kidney transplantation.6
Initial approach of aHUS requires red blood cells transfusion and hemodialysis, which were promptly initiated in this case. Platelet transfusion is contraindicated despite thrombocytopenia, except in cases of active bleeding, since it may exacerbate TMA.6 Plasma therapy is the classical first choice treatment for all cases of aHUS.1,2,6 Plasmapheresis is prefered to plasma infusion because it assures the delivery of functional complement regulatory proteins and the removal of dysfunctional factors, with lower risk of volume overload. Should be initiated as early as possible, with exchange of 1.5 plasma volume per session, and continued until platelet count, hemoglobin, and LDH levels normalization.1,5–7
Plasmapheresis has been the cornerstone of aHUS treatment since 1980 and was the only therapy available until 2010. A recent therapeutic alternative is eculizumab – a humanized monoclonal IgG antibody which binds to the C5 complement protein preventing the formation of C5b-9 (membrane attack complex) – already approved by the EMA and FDA.1,2,4,6 Two prospective phase II trials have demonstrated its efficacy on improving the platelet count and renal function of aHUS patients, including those with progressing TMA and those with long-standing, substantial kidney damage.8 A posterior analysis confirmed the persistence of the clinical benefits after two years of follow-up.9 Eculizumab was also associated with significant improvements in health-related quality of life.8
Barriers to eculizumab use include patient safety concerns, as infection with encapsulated bacterial organisms and the elevated cost.2 On the other hand no cumulative toxicity or unexpected serious infections were observed, and survival was 100% on pivotal prospective studies,8–10 turning Eculizumab into a very promising option for aHUS patients treatment.
FundingNone.
Conflict of interestNone.

