



INTRODUCCIÓN
A pesar de los enormes avances en el reconocimiento, el diagnóstico y el tratamiento de los pacientes críticos, la tasa de mortalidad en esta población sigue siendo muy elevada. En los últimos años se ha centrado la atención en el papel de la hemostasia y, en particular, de los trastornos hemostáticos1.
La hemostasia es el conjunto de procesos que tiene por misión dos funciones bien diferentes: por una parte, detectar las pérdidas de continuidad que aparezcan en el árbol vascular y detener las pérdidas de sangre que se pudieran producir, y por otra, mantener la permeabilidad de los vasos. La hemostasia fisiológica es el resultado de un fino balance entre los sistemas de las enzimas proteolíticas, los cofactores y los inhibidores que concurren en espacio y tiempo2,3. Comprende un elevado número de complejas interacciones entre los componentes de la sangre y las paredes de los vasos sanguíneos. En esquema las podríamos reunir en 5 grupos diferentes cuya activación presenta, en ocasiones, fases comunes. Éstas son: una vasoconstricción local, las reacciones de adhesión y liberación plaquetaria, la formación de fibrina y la subsiguiente estabilización del trombo plaquetario y, finalmente, la eliminación de este trombo por medio de los mecanismos fibrinolíticos4,5.
El trombo plaquetario es capaz de detener las hemorragias que se producen en los capilares y las pequeñas vénulas; sin embargo, en caso de vasos de mayor calibre o con una presión intravascular alta, como en el árbol arterial, la firmeza de dicho trombo es insuficiente para detener la hemorragia. En estos casos, las plaquetas deben ser estabilizadas con una malla de fibrina.
La trombina es la responsable de la transformación del fibrinógeno soluble en fibrina insoluble, que polimerizará y formará la malla necesaria para la estabilización del trombo plaquetario. La formación de trombina es el punto final de una serie de reacciones enzimáticas, en las cuales una proenzima es convertida en enzima activa que, a su vez, activará otra proenzima. Este modelo de activación se ha descrito como "en cascada" y constituye el eje central de la coagulación de la sangre.
La principal función de la fibrina es contribuir al taponamiento de la lesión a través de la cual sangra un vaso. Sin embargo, tan pronto como el objetivo se consigue, deben existir mecanismos que limiten su formación. Si estos mecanismos no funcionan adecuadamente aparece el riesgo de trombosis (fig. 1).
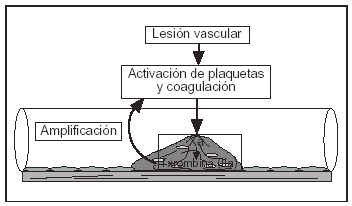
Figura 1. Situación procoagulante en la trombólisis.
El sistema fibrinolítico es el encargado de eliminar la fibrina del lecho vascular, además de intervenir en otros importantes fenómenos biológicos. La lisis de un trombo se inicia cuando el activador del plasminógeno tisular (t-PA) es liberado por el endotelio. Un factor que activa su liberación es la estasis proximal a la trombosis de los vasos. Para que el t-PA liberado sea activo es necesario que se fije a la fibrina del trombo. Posteriormente, el t-PA actuará sobre el plasminógeno, convirtiéndolo en plasmina que digerirá la fibrina.
Este proceso localizado en el trombo está sujeto a una estrecha regulación. La potencia del sistema coagulativo y la existencia en el mismo de mecanismos de retroalimentación aseguran la eficacia hemostática, pero también conllevan el riesgo de activación más allá de las necesidades de ésta, dando lugar al fenómeno trombótico. Esto hace necesaria la existencia de mecanismos biológicos que limiten y modulen dicha activación, la ausencia de los cuales sería incompatible con la vida6. Por otra parte, el sistema fibrinolítico, que actúa como regulador del sistema coagulativo eliminando la fibrina innecesaria para la hemostasia, precisa, a su vez, de un sistema de control que evite su activación excesiva y, con ella, la hemorragia6.
En síntesis, los recientes avances en el conocimiento de la coagulación sanguínea no han permitido establecer qué aspectos son incorrectos en el modelo clásico de la activación de la coagulación en "cascada", con 2 vías, extrínseca e intrínseca, que convergen en el factor X. Sin embargo, aunque esto en realidad nunca ha tenido una repercusión en la práctica clínica, hoy día parece demostrado el papel central del factor VII como iniciador de la coagulación in vivo al formar un complejo con el factor tisular (FT), con lo que se establece la activación, tanto del factor X como del factor IX7,8.
Obviamente, la sospecha clínica es evidente cuando existe un sangrado activo (úlcera, gastrointestinal, traumatismo), pero a menudo el sangrado es mínimo o las modificaciones de la coagulación se evidencian por las alteraciones de las pruebas de laboratorio, y su relevancia clínica es cuestionable9.
Por otra parte, la incidencia de sangrado grave e incontrolado con escasas o nulas alteraciones de la coagulación no es desdeñable en nuestros pacientes, tanto en el momento del ingreso como en el curso evolutivo, y se ha descrito una incidencia de hemorragia de hasta el 40% en los pacientes traumáticos que fallecen.
Revisaremos el pro y el contra de la terapia prohemostática disponible y destacaremos los resultados con análogos de la lisina, de la desmopresina y del factor VII recombinante activado (rFVIIa).
ALTERACIONES DE LA COAGULACIÓN
Tras la agresión se desencadena una serie de respuestas orgánicas programadas que tienden a limitar el cuadro inflamatorio. La pérdida de control local induce una respuesta inflamatoria generalizada, rápida y ampliada, controlada humoral y celularmente (complemento, cininas, coagulación y cascada fibrinolítica) y desencadenada por activación conjunta de los fagocitos y las células endoteliales. Si esta respuesta inflamatoria no es adecuadamente modulada, se origina un síndrome de respuesta inflamatoria sistémica (SIRS)10.
Básicamente, lo que ocurre y se ha demostrado de manera amplia en modelos animales con cuadros clínicos inducidos que caracterizan las diversas fases de la sepsis según las definiciones de la ACCP/SCCM, es que existe una rotura del equilibrio hemostático en sentido protrombótico. Esta rotura está mediada por una serie de alteraciones que se producen en la compleja red de citocinas, moléculas necesarias para garantizar la capacidad de respuesta del organismo ante las agresiones externas, pero que a las concentraciones no fisiológicas en las que aparecen en estos cuadros son capaces de generar una serie de mecanismos que contribuirán no sólo a la generación de fibrina, sino a una respuesta inflamatoria desmedida11. La consecuencia es la formación de fibrina (la situación clínica más extrema sería la coagulación intravascular diseminada [CID]), que se deposita en la microcirculación de diversos órganos y contribuye así a la presentación del síndrome de disfunción multiorgánica. La fibrinolisis, todavía eficaz en una fase precoz, se inhibe progresivamente, lo que agrava el desequilibrio y favorece la formación de microtrombos1. Incluso la anticoagulación endógena, constituida por la antitrombina III (AT-III) y por el sistema proteína C-proteína S (PC-PS), experimenta una inhibición progresiva debida al consumo excesivo. Además, hay una inhibición de la formación de glucosaminoglucanos endógenos y, por tanto, de la formación de complejos trombina-antitrombina (TAT) en cuanto que la noxa (endotoxina) inhibe la trombomodulina (activador de la PC) y estimula la C4bBP (proteína de fase aguda que se une a la PS y la inactiva)12. Además, hay que tener en cuenta el importante papel de las citocinas proinflamatorias (factor de necrosis tumoral [FNT], que es el principal responsable de la inhibición de la fibrinolisis13, e interleucinas (JL) 1 y 6, que median los trastornos de la coagulación) que, como veremos más adelante, intervienen en la activación de la coagulación.
Asimismo, la trombocitopenia es una coagulopatía frecuente en los pacientes críticos cuyo mecanismo es la producción de hemofagocitosis por agregación sistémica y, en consecuencia, por consumo12.
Aunque este mecanismo es extrapolable, en un grado más o menos intenso de activación de la coagulación, a los distintos síndromes que determinan el espectro de paciente crítico, la alteración hemostática mejor conocida es la CID secundaria a la sepsis, en la que la activación de la coagulación se produce a través de la vía extrínseca al unirse el factor tisular (FT) a su cofactor, el factor VII (a)14-16.
EL FT está presente en la membrana plasmática de diversos tipos celulares, aunque en circunstancias normales no se expresa en los monocitos y en las células del endotelio. Sin embargo, su expresión en estas células puede ser inducida, durante el curso de una infección, por endotoxinas bacterianas y citocinas, como el FNT17-19. Una vez expuesto en la cara externa de la membrana plasmática, el FT inicia una actividad procoagulante localizada que puede dar lugar, en último término, a la formación de un coágulo20,21.
El FT es una glucoproteína de membrana que activa el inicio de la cascada de las serinproteasas del proceso de la coagulación. Una vez expuesto en la superficie celular, el FT actúa como receptor de alta afinidad del factor VII del plasma. El complejo formado por el FT y el factor VII, bien en forma de cimógeno o en forma activada (FVIIa), o sea, FT-FVII o FT-FVIIa por la unión al receptor, activa al factor X, tanto directamente como mediante la activación del factor IX. El factor Xa es el componente activo del complejo de la protrombinasa (Xa-Va) que convierte la protrombina en trombina (IIa), que es la que participa en los procesos de activación plaquetaria. Éstos culminan en la formación de un trombo plaquetario y promueven la transformación del fibrinógeno en una malla de fibrina polimerizada que estabiliza dicho trombo14.
El gen FT humano se localiza en la posición p21-22 del cromosoma 1, donde ocupa un total de 12,4 kb organizadas en 6 exones y 5 intrones22,23. La proteína originada está constituida por 3 dominios: una pequeña región intracitoplasmática, una zona hidrófoba de anclaje en la membrana celular y una región hidrófila de mayor tamaño orientada hacia el exterior de la célula19. El FT pertenece a la familia de los receptores de las citocinas y muestra una mayor semejanza con los receptores tipo II, como los del interferón alfa y gamma.
En condiciones normales, el FT está presente en algunas células pero ausente en muchas otras. En general, en los tejidos humanos se distribuye a modo de envoltura del organismo frente al exterior24 y es más abundante en la epidermis que en la dermis. En el tracto digestivo, tanto en la mucosa como en la capa muscular, también existe una elevada expresión de FT, al igual que en el pulmón, el sistema nervioso central (SNC) y el miocardio24.
El FT no se expresa normalmente en la pared vascular, a excepción de una expresión moderada en la adventicia (este patrón evita la activación espontánea de la coagulación en el espacio intravascular y previene hemorragias que puedan suceder tras una lesión vascular)14.
Sin embargo, tanto en los monocitos como en las propias células endoteliales, la expresión del FT puede ser inducida por la exposición a endotoxinas bacterianas, ésteres de forbol, lectinas, factores de crecimiento y mediadores celulares de la respuesta inmunológica (linfocitos T)17-19.
De todas maneras, la presencia de FT en la superficie celular no basta para que actúe como inductor de la coagulación, ya que es necesario que se produzca una remodelación de la bicapa lipídica de la membrana plasmática que propicia la unión de la región hidrófoba de la glucoproteína a residuos lipídicos de fosfatidilcolina y fosfatidilserina25,26; y es que el FT en presencia de fosfolípidos aumenta espectacularmente su afinidad por el FVII (a), dando lugar a la formación de complejos estables FT-FVII (a) sobre la superficie fosfolipídica20,24,26 que resultan mucho más eficaces que la forma libre de FVII en la activación de los factores IX y X de la coagulación27-29.
Hoy día se cree que los depósitos de fibrina son consecuencia de la activación de la coagulación, mediada por la unión del FT al FVIIa, y de la disfunción del sistema anticoagulante, manifestada por la disminución de los valores de AT-III, proteína C y proteína S. Además, la deposición de fibrina puede verse favorecida por la inhibición, en primera instancia, del sistema fibrinolítico, que se manifiesta por un aumento de las concentraciones del inhibidor del activador tisular del plasminógeno (PAI), aunque en una segunda fase, el sistema fibrinolítico puede resultar activado y dar lugar también a complicaciones hemorrágicas16. Obviamente, también existe un mecanismo de control de esta activación de la coagulación, el inhibidor plasmático del FT (IPFT), que es producido por las células endoteliales y los megacariocitos y que, al unirse al complejo FT-FVIIa, inactiva su acción catalítica.
La progresión de la activación de la coagulación tras la formación del complejo FT-FVIIa depende fundamentalmente de la capacidad de los inhibidores naturales de la coagulación, AT-III, PC y PS, cuyas concentraciones son bajas y el grado de reducción esta íntimamente relacionado con el pronóstico del síndrome30.
En los procesos sépticos, el sistema de coagulación y las plaquetas están fuertemente activados, tanto en los cuadros complicados como en los no complicados, si bien en los segundos se objetiva un descenso del factor XII, mientras que en los primeros hay depleción de todos los factores de la coagulación, consumo de plaquetas y prolongación de los tests globales de la coagulación, lo que indica una coagulopatía de consumo31.
Además del mecanismo fisiopatológico ampliamente documentado de activación de la vía extrínseca de la coagulación, debemos considerar que, en presencia de proliferación y activación de monocitos y macrófagos que destruyen las células hematopoyéticas, se produce hemofagocitosis. Ésta sería responsable de la trombopenia que observamos con frecuencia en los pacientes críticos, especialmente en los sépticos, que no está determinada por factores como CID, hemorragias y transfusiones, terapias (incluida la heparina) y/o enfermedades sanguíneas. En la génesis de esta hemofagocitosis desempeña un papel muy importante, junto con las citocinas inflamatorias, el factor estimulante de colonias de macrófagos (M-SCF)32.
En resumen, en los pacientes críticos existe un desequilibrio o desregulación entre las actividades procoagulantes y las anticoagulantes. El endotelio vascular, en condiciones normales, regula la formación del coágulo y su lisis. Sus funciones pueden estar alteradas por la exposición a agentes infecciosos, a toxinas o a mediadores inflamatorios, como la endotoxina bacteriana, los componentes del complemento, la IL-1, el FNT o las proteasas granulocitarias17,24. El endotelio lesionado o estimulado manifiesta funciones procoagulantes: puede expresar FT33 (que se expresa constitutivamente en tejidos que no están expuestos a la sangre, como la adventicia de los vasos sanguíneos, la piel, el tejido nervioso, el glomérulo renal, el líquido amniótico, etc.), disminuir la trombomodulina, liberar el PAI, el factor activador de las plaquetas, la endotelina y el factor von Willebrand y disminuir la síntesis de óxido nítrico34. La exposición del subendotelio a la sangre produce adhesión y agregación plaquetaria, así como la puesta en marcha de los mecanismos de coagulación. La endotoxina incrementa la adhesión de granulocitos y monocitos al endotelio capilar y causa una lesión endotelial difusa, mediada por IL-1 y FNT17,24,35. Probablemente, la expresión del FT en los monocitos es decisiva para desencadenar una activación del sistema de la coagulación. La prevalencia de las actividades procoagulantes finalmente causará la conversión del fibrinógeno en monómeros de fibrina y la formación de fibrina soluble. Ésta puede eliminarse de la circulación o ser lisada por la acción del sistema fibrinolítico activado. Sin embargo, la fibrina soluble puede quedar confinada en determinadas áreas y su polimerización puede ser mediada por la lesión endotelial o por reacciones vasomotoras, aún poco clarificadas. La fibrina soluble y los trombos microvasculares pueden ser lisados por el sistema fibrinolítico si las actividades fibrinolíticas están suprarreguladas; sin embargo, si están inhibidas, los microtrombos ricos en fibrina pueden persistir, lo que resulta en una lesión endotelial, una lesión hística y, finalmente, en insuficiencia orgánica.
Kauffman et al profundizaron en esta idea y llevaron a cabo autopsias muy detalladas a 16 pacientes fallecidos a causa de un traumatismo craneoencefálico grave y afectados de coagulación intravascular diseminada biológica y disfunción multiorgánica, con el objeto de hallar microtrombos diseminados36,37. A pesar de que la necroscopia sistemática no reveló la presencia de lesiones sugestivas de CID en estos pacientes, un minucioso examen posterior demostró la presencia de microtrombos en el 88% de los cadáveres (fundamentalmente localizados en el cerebro, el pulmón y el hígado) y petequias en la superficie cerebral en 10 de 16 pacientes. Con estos datos, los autores demostraron que la correlación anatomopatológica de CID respecto a los datos clínicos era muy superior a la que se suponía hasta ese momento, que la prevalencia de CID era mayor de la esperada en pacientes fallecidos por traumatismo craneoencefálico y que la microtrombosis diseminada con disfunción multiorgánica concomitante era la causa de estas muertes, superando a los fenómenos hemorrágicos.
¿QUÉ PRUEBAS DIAGNÓSTICAS EMPLEAMOS PARA DETECTAR ALTERACIONES DE LA COAGULACIÓN Y CUÁNDO LAS REALIZAMOS?
Como ya hemos mencionado con anterioridad, un serio problema para interpretar la prevalencia de coagulopatía en los pacientes críticos se deriva de los diferentes criterios diagnósticos utilizados en los distintos trabajos que analizan este tema, sin que se cite claramente ninguna gradación (activación de la coagulación, CID y CID clínica) ni se definan los parámetros de la coagulación con un mayor poder predictivo ni el momento del cuadro en que deben ser analizados.
La revisión de la bibliografía hace que podamos considerar como pruebas de laboratorio princeps los siguientes parámetros:
Concentración de antígeno de FT, que realmente está midiendo la concentración de FT capaz de activar la vía extrínseca de la coagulación.
Fragmento 1 + 2 de protrombina, que mide indirectamente el grado de formación de trombina.
Complejo TAT, que indica la inhibición de la trombina por parte de la AT-III.
Fibrinopéptido A, que es un péptido que se desprende del fibrinógeno durante su transformación en fibrina y que indica la formación de trombina.
Dímeros d (DD), indicativos de fibrinolisis.
AT-III, que es un anticoagulante endógeno inhibidor de la trombina.
Algunos autores39,40 han considerado CID clínica cuando los pacientes presentaban cifras de fibrinógeno inferiores a 130 mg/dl y PDF superiores a 40 ng/ml, mientras que otros utilizan complicados algoritmos diagnósticos que incluyen hasta 6 parámetros41.
Nossel et al42, en un estudio realizado tras la inducción de abortos en embarazadas y según la experiencia clínica del tratamiento de pacientes con CID, establecen varios estadios: estadio I, caracterizado por valores elevados de productos de degradación del fibrinógeno (PDF) o DD; estadio II, definido por la prolongación del TP o del TTPa, o por un descenso del 50% del valor de base del fibrinógeno. En la fase final, los pacientes presentan hipofibrinogenemia (< 100 mg/dl) y trombocitopenia. En estos pacientes también se observan concentraciones disminuidas de antitrombina III, plasminógeno y α2-antiplasmina. Los fenómenos hemorrágicos o de trombosis pueden ocurrir en cualquier estadio. El estadio III de la CID se acompaña de una mortalidad muy elevada.
Los cambios más tempranos detectados en los controles analíticos sistemáticos de la coagulación son los valores elevados del complejo plasmina-PDF o DD. Un valor elevado de los DD es un marcador más sensible y específico de la degradación de la fibrina que la determinación de los PDF. Algunos investigadores han recomendado determinar ambos, los PDF y los DD, para realizar el diagnóstico de CID. Greenberg et al43 hallaron una buena correlación entre ambas determinaciones. En general, no está indicado determinar los PDF en los pacientes con sospecha de CID, lo que se reserva para los pacientes con una prueba negativa de DD.
La medición del DD determina la degradación de fibrina, mientras que los PDF no permiten distinguir entre la degradación de fibrinógeno y la de fibrina. Además, la determinación de PDF requiere unos tubos especiales para recoger la muestra, y existen resultados falsos positivos producidos por la heparina y otros factores que inhiben la formación de la fibrina en el plasma del paciente. La determinación de los DD se puede realizar sin la necesidad de utilizar tubos especiales, gracias a una técnica de anticuerpos monoclonales. En contraste, la determinación de PDF actuales se basa en técnicas de anticuerpos policlonales, tanto tanto al fibrinógeno como a los fragmentos D.
En el estudio de Greenberg et al43, los valores de DD superiores a 10 ng/ml con frecuencia están asociados a hipofibrinogenemia (< 100 mg/l) y trombocitopenia. La mayoría de los pacientes críticos tendrán valores de DD superiores a 1-2 μg/ml debido a que es un marcador sensible de la formación de fibrina intravascular. Sin embargo, hay que tener en cuenta las limitaciones de la determinación de DD, ya que tienen una vida media de tan sólo 3,5 h. Actualmente se dispone de técnicas de laboratorio para realizar determinaciones semicuantitativas de DD, útiles en el diagnóstico de la enfermedad tromboembólica44, que están facilitando su determinación en los pacientes críticos.
La determinación del fragmento 1.2 nos indica la actividad del complejo protrombinasa, que puede ser un marcador fiable de la actividad de la coagulación. La observación del fragmento 1.2 en varios pacientes con sospecha de CID demostró que sus valores disminuían cuando la enfermedad de base mejoraba44. Los valores de DD a menudo permanecían elevados y disminuían más lentamente que los valores del fragmento 1.2, lo que demuestra que la fibrinolisis puede continuar después de que la formación de fibrina haya sido inhibida.
Los pacientes afectados de CID presentan un aumento de los complejos trombina-factor Xa-antitrombina III, así como una disminución de los valores plasmáticos de antitrombina III por su consumo y degradación por la elastasa.
En resumen, hoy día no existen pruebas de laboratorio fácilmente reproducibles para observar directamente la activación de la coagulación sanguínea28, hecho que dificulta enormente tanto su diagnóstico como su seguimiento; no obstante, parece claro que todos los parámetros "clásicos" (alargamiento del tiempo de protrombina [TP] y tiempo de tromboplastina parcial activado [TTPA], descenso en el recuento de plaquetas y fibrinógeno, y aumento de PDF y DD), que conducen al diagnóstico son normales o sólo están ligeramente alterados en las formas iniciales de activación de la coagulación; en el diagnóstico de las formas subclínicas está especialmente indicado evaluar la TAT, el fibrinopéptido A, el fragmento 1 + 2, el DD, el PDF y el complejo plasmina-antiplasmina (PAP)46.
Así, para diagnosticar una activación de la coagulación mediante parámetros de laboratorio debemos encontrar datos que evidencien47,48:
1. Una actividad procoagulante: evaluada por elevación de fragmento 1 + 2 de la protrombina, del fibrinopéptido A, del fibrinopéptido B, del complejo TAT y del dímero D.
2. Una actividad fibrinolítica: evaluada por elevación del dímero D, de los PDF, de la plasmina y del complejo PAP.
3. El consumo de inhibidores: evaluado por el descenso de los valores de AT-III, de la α2-antiplasmina, del heparincofactor II, de los valores de PC y PS y elevación de los complejos TAT y PAP.
4. Fallo orgánico: evaluado por elevación de LDH y de la creatinina, y descenso del pH y de la PaO2.
Para el diagnóstico de activación de la coagulación es necesario encontrar una anormalidad de cada grupo (I, II, III) y dos del grupo IV.
Obviamente, debemos encontrar una manera de clasificar, de forma comparativa entre distintos centros, los diferentes grados de activación de la coagulación, y aunque no es fácil por falta de estudios, el sistema de puntuación establecido por Bick (1995), conjuga los 4 grupos de test de laboratorio con parámetros clínicos y hemodinámicos (tabla 1). Esto permite establecer una activación de la coagulación subclínica o inapreciable (score > 90 puntos), una activación discreta (score 75-89), una activación moderada (score 50-74) y grave (score < 49)48,49.
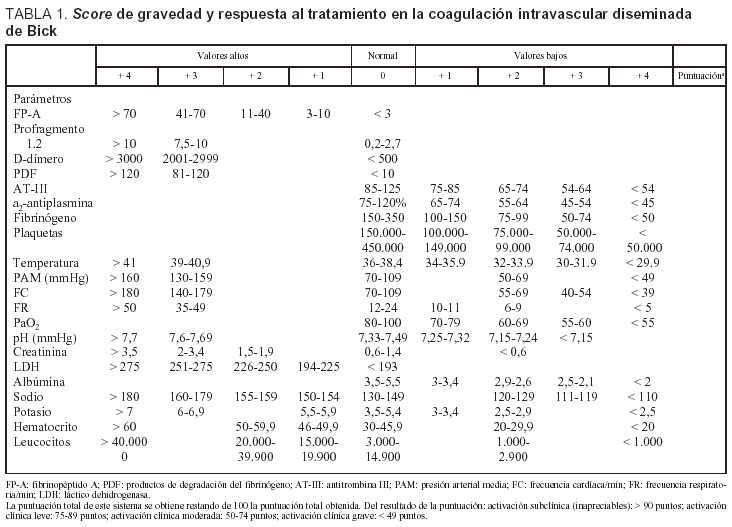
Aunque poco introducidos en nuestro entorno, también deberíamos considerar los criterios diagnósticos de CID propuestos por Aoki (1988) y aceptados por el Ministerio de Salud del Japón (tabla 2), que aportan sencillez respecto a los de Bick aunque no son aplicables en neonatos, embarazadas ni en la hepatitis fulminante. Según estos criterios, se consideraría la existencia de CID si la puntuación es ≥ 7, sospecha de CID si es igual a 6 y baja probabilidad de CID si es ≤ 5, con un score diferente si estamos ante pacientes con leucemia, anemia aplásica o postratamiento con antineoplásicos (se considera un score de 0 en el recuento de plaquetas y en los síntomas de hemorragia)50-52.

¿ES LA ALTERACIÓN DE LA COAGULACIÓN EN EL PACIENTE CRÍTICO UNA SITUACIÓN DEPENDIENTE?
Síndrome de coagulación intravascular diseminada
No debe entenderse como un diagnóstico clínico, sino como la expresión clínica máxima de la respuesta biológica a un estímulo patológico. La activación persistente del sistema de la coagulación como consecuencia de una enfermedad de base produce un menor o mayor grado de coagulación intravascular diseminada, que desaparecerá una vez cese el estímulo.
Como ya hemos comentado anteriormente, la identificación de los factores que inducen una CID es esencial para plantear una estrategia terapéutica adecuada, y la lesión vascular que estimula la activación del sistema de complemento y de los factores de la coagulación, así como el contacto del TF con la sangre, es el factor desencadenante principal. La importancia de estos mecanismos no está bien definida debido a la dificultad para disponer de métodos para cuantificar su activación.
La naturaleza dinámica de los cambios de las proteínas de la coagulación en una CID ha sido extensamente descrita en un trabajo clásico de Nossel et al42 tras la inducción de abortos en embarazadas. Cuando existe un estímulo que pone en marcha la formación de trombina, mientras éste no cese la formación continúa existiendo una activación y un consumo de las proteínas de la coagulación, y se forman depósitos microvasculares de fibrina que el organismo trata de lisar generando plasmina. Ésta causa una mayor degeneración de los factores V y VIII y de fibrinógeno. Las plaquetas se activan y su vida media se acorta por el proceso de coagulación. Existirá también una degradación del fibrinógeno y de la antitrombina III a causa de la elastasa liberada por los neutrófilos durante la coagulación sanguínea. En un intento por regular la formación de trombina tendrán lugar fenómenos como los complejos formados por la trombina y el factor Xa con la antitrombina III y la reducción de los valores circulantes de proteína C. El resultado final será una depleción de la mayoría de las proteínas de la coagulación y de sus inhibidores, así como una reducción del número de plaquetas. Con frecuencia, este hecho produce graves problemas hemorrágicos y trombóticos. El grado de activación y consumo de las plaquetas, las proteínas de la coagulación, los inhibidores de la coagulación y la fibrinolisis dependerá de la intensidad y la duración del estímulo procoagulante45.
Existen 2 cuadros clínicos bien diferenciados como expresión de una CID53,54. El primero es una diátesis hemorrágica con petequias, equimosis, hematuria y hemorragia gingival, gastrointestinal y del sistema nervioso central. Es característica la presencia de hipofibrinogenemia y trombocitopenia. Sin embargo, algunos pacientes presentan una inhibición funcional plaquetaria más profunda debido a la liberación del contenido de sus gránulos y cuerpos densos. La segunda forma de presentación de una CID es la trombosis. En una CID aguda es frecuente observar complicaciones hemorrágicas y trombóticas simultáneamente55.
Síndrome de distrés respiratorio agudo (SDRA)
Es un cuadro de insuficiencia respiratoria aguda progresiva que puede aparecer en pacientes con sepsis, multitransfundidos, politraumatizados, con pancreatitis agudas postoperados, etc. La mortalidad global es superior al 50%, aunque depende de la etiología responsable56. En el SDRA existe un trastorno de la permeabilidad capilar pulmonar que ocasiona un paso de células y proteínas al espacio alveolar debido a un proceso inflamatorio local en el que intervienen diversos factores celulares y humorales. Los neutrófilos y monocitos liberan enzimas, citocinas y eicosanoides que lesionan el endotelio capilar pulmonar y aumentan la permeabilidad. En la etiopatogenia intervienen las citocinas proinflamatorias IL-1 y FNT58-60 que, como ya hemos mencionado, favorecen una situación clínica de shock, edema pulmonar, diátesis hemorrágica, isquemia intestinal, necrosis tubular renal, hemorragia adrenal y muerte. En este cuadro se produce una disregulación con activación del sistema de la coagulación; también se encuentra trombosis en la microcirculación y en ocasiones en vasos de mayor tamaño32,60, y en las determinaciones analíticas se observan cifras elevadas de FT60, del fragmento 1 + 2 de la protrombina y de dímero D, que se han relacionado con una mala evolución clínica63.
Estos microtrombos podrían ocluir los pequeños vasos o causar agregación y adhesión plaquetarias, con la liberación concomitante de sustancias vasoconstrictoras, como el tromboxano A264. Todo ello se sustenta en una base experimental sólida, dado que se hallaron resultados similares en experimentos de microembolización en animales, fundamentalmente en perros65,66. Diversos estudios previos habrían demostrado una estrecha relación entre los valores de PDF en diversas situaciones clínicas y la aparición de SDRA67-69. En otros trabajos se ha descrito un incremento de la capacidad procoagulante del líquido alveolar70 y un déficit en la actividad fibrinolítica de este líquido72,73, con ausencia de urocinasa (habitual en esta localización) inhibida posiblemente por valores muy altos de su inhibidor fisiológico (PAI-1), así como un incremento en la expresión de FT sobre el endotelio y los monocitos74.
En definitiva, la obstrucción generalizada de los capilares pulmonares debida a microtrombos de fibrina y plaquetas con la liberación de sustancias vasoconstrictoras parece ser uno de los sustratos etiopatogénicos del SDRA.
Traumatismo craneoencefálico
Las alteraciones de la coagulación son relativamente comunes en los pacientes con traumatismo craneoencefálico (TCE) (10-18%) y más en los TCE graves (25-40%). Estas alteraciones, que se relacionan con un mal pronóstico41,75, son posiblemente debidas a la formación de microtrombos como consecuencia de la aparición en el torrente vascular de FT procedente del tejido encefálico, rico en FT, que es una proteína que se halla en concentraciones elevadas en el cerebro76 y que provoca un proceso de activación generalizada y descontrolada de la coagulación, con la formación de grandes cantidades de trombina, consumo de plaquetas, fibrinógeno y factores de la coagulación, trombosis microvascular, activación concomitante del sistema fibrinolítico y, en definitiva, CID. No es extraño que una acumulación tan notable de "poder procoagulante" sea capaz, en una situación patológica, de extralimitar la respuesta hemostática de manera anómala lejos del territorio lesionado77.
Algunos autores han postulado que la masiva liberación de catecolaminas que experimentan los pacientes con TCE puede exacerbar la coagulopatía inherente a estos individuos, estimulando la activación plaquetaria y aumentando el daño endotelial78. A este respecto, Kearney et al79 iniciaron en 1989 un estudio prospectivo en 36 pacientes con TCE grave, con una puntuación < 10 en la Escala de Coma de Glasgow, a los que compararon con controles sometidos a neurocirugía electiva en cuanto a los valores seriados en orina de normetanefrina y ácido vanililmandélico, además de diversos parámetros de la coagulación79. Se comprobó que los valores de catecolaminas de los primeros eran significativante mayores. Para los autores, este dato es un factor determinante para comprender por qué los pacientes traumáticos suelen presentar CID, a diferencia de los pacientes sometidos a neurocirugía, a pesar de que es habitual en la práctica neuroquirúrgica la sección de importantes zonas de tejido cerebral y, por consiguiente, el tránsito inevitable de material tromboplastínico al torrente circulatorio. El importante incremento de catecolaminas del paciente traumático puede ser una de las vías etiopatogénicas de la CID en el TCE, al provocar una intensa activación plaquetaria80.
Otros procesos
Aunque hemos hecho referencia a los principales procesos en los que parece estar más claro el mecanismo patogénico de afección de la coagulación, es posible encontrarlo en los pacientes críticos con independencia del mecanismo desencadenante. Así, en los pacientes en los que el aporte de oxígeno tisular es insuficiente para mantener una adecuada reserva energética celular se genera una necrosis tisular y las células liberan factor tisular que, junto con la lesión del endotelio vascular debida a un aporte insuficiente de oxígeno, son responsables del inicio de la actividad procoagulante.
En resumen, y en líneas generales, se puede esperar un bajo grado de activación de la coagulación en las enfermedades cardiovasculares y autoinmunes, en las alteraciones vasculorrenales, hematológicas e inflamatorias y un alto grado en los accidentes obstétricos (embolia de líquido amniótico, abrupción placentaria, síndrome de feto retenido, eclampsia, aborto), hemólisis intravascular (reacciones hemolíticas postransfusionales, transfusiones masivas, hemólisis menores), septicemia (gramnegativos-endotoxina) y positivos (mucopolisacáridos), viremias (virus de la inmunodeficiencia humana, hepatitis, varicela, citomegalovirus), leucemia (aguda promielocítica y mielocítica), quemaduras, traumatismos, necrosis masiva de tejidos, lesiones vasculares, lesiones por aplastamiento y enfermedades hepáticas agudas (fallo hepático fulminante, obstrucción biliar)52.

