




Determinar la frecuencia y duración de episodios de despolarización cortical propagada (CSD y CSD-like) en pacientes con lesiones por traumatismo craneoencefálico (TCE) e infartos malignos de la arteria cerebral media (IMACM) que requirieron craneotomía.
DiseñoEstudio descriptivo-observacional desarrollado durante 19 meses.
ÁmbitoPacientes neurocríticos.
PacientesEstudio realizado en 16 pacientes (9 IMACM y 7 TCE graves) que requirieron tratamiento quirúrgico.
IntervencionesColocación de una tira de 6 electrodos en el córtex perilesional para el registro continuo de electrocorticografía (ECoG).
Variables de interés principalesEn todos los casos se determinó la hora, el número y la duración de los episodios de CSD y CSD-like detectados.
ResultadosDe los 16 registros de ECoG analizados, 9 presentaron fenómenos CSD y CSD-like, de duración y frecuencias muy variables.
ConclusionesLos episodios de CSD y CSD-like se detectan con frecuencia en el córtex en las regiones de penumbra isquémica y/o traumática de pacientes con un IMACM que han requerido craniectomía descompresiva o afectos de un TCE con contusiones cerebrales.
To determine the frequency and duration of cortical spreading depolarization (CSD) and CSD-like episodes in patients with traumatic brain injury (TBI) and malignant middle cerebral artery infarction (MMCAI) requiring craniotomy.
DesignA descriptive observational study was carried out during 19 months.
SettingNeurocritical patients.
PatientsSixteen patients were included: 9 with MMCAI and 7 with moderate or severe TBI, requiring surgical treatment.
InterventionsA 6-electrode subdural electrocorticographic (ECoG) strip was placed onto the perilesional cortex.
Main variables of interestAn analysis was made of the time profile and the number and duration of CSD and CSD-like episodes recorded from the ECoGs.
ResultsOf the 16 patients enrolled, 9 presented episodes of CSD or CSD-like phenomena, of highly variable frequency and duration.
ConclusionsEpisodes of CSD and CSD-like phenomena are frequently detected in the ischemic penumbra and/or traumatic cortical regions of patients with MMCAI who require decompressive craniectomy or of patients with contusional TBI.
El ictus y el traumatismo craneoencefálico (TCE) constituyen causas de discapacidad funcional importante. Las lesiones tardías (secundarias) y no las inmediatas (primarias) son las que modifican de forma más significativa el pronóstico funcional de estos pacientes. En ambos casos, el objetivo terapéutico en la fase aguda debe ir dirigido a rescatar el tejido potencialmente recuperable, correspondiente a las áreas de penumbra.
Las áreas de penumbra isquémica se definen como áreas de tejido cerebral perilesional estructuralmente intactas pero con graves alteraciones metabólicas, y en el contenido del espacio intra y extracelular1. Este tejido es especialmente sensible y se deteriora ante cualquier acontecimiento adverso sobreañadido. Los esfuerzos terapéuticos en las últimas 2 décadas en el paciente neurocrítico se han dirigido a prevenir, detectar y tratar de forma rápida los acontecimientos adversos que pueden hacer que las zonas de penumbra progresen a áreas de tejido cerebral dañado de forma irreversible por fenómenos de apoptosis y/o de necrosis.
En los últimos años algunos autores han rescatado el concepto de despolarización propagada o cortical spreading depression (CSD) y los fenómenos denominados CSD-like como una de las causas más frecuentes y menos estudiadas del deterioro metabólico del tejido cerebral en el paciente neurocrítico2–5 y en especial en pacientes con un TCE y con un ictus isquémico2,3. El fenómeno denominado CSD fue descrito inicialmente por Leão en 1944 al intentar inducir crisis epilépticas mediante estimulación eléctrica en el córtex frontal del conejo6,7. Este fenómeno eléctrico se caracteriza por una despolarización de la actividad sináptica y por la propagación de la onda de despolarización por la sustancia gris cortical acompañada de una redistribución masiva de los iones a nivel intra y extracelular. La característica fundamental de este fenómeno es su propagación por el neocórtex en forma de onda expansiva lenta8. Experimentalmente, la CSD es una respuesta inespecífica del córtex a un gran número de estímulos (isquemia, anoxia, traumatismos o infusión de potasio o glutamato)8. Aunque la CSD afecta a todo el sistema nervioso central, tiene una especial preferencia por el hipocampo y en especial por la región CA18,9.
En el electrocorticograma (ECoG) la CSD se detecta por una depresión súbita de la amplitud del registro eléctrico que se propaga a una velocidad de 1-5mm/min. Aunque en la literatura sobre el tema hay todavía una cierta confusión en la taxonomía, existe coincidencia en distinguir entre 2 fenómenos claramente diferenciados: la CSD clásica y los fenómenos CSD-like, también denominados despolarizaciones periinfárticas (PID, del inglés peri-infarct depolarization).
La CSD clásica (CSD normóxica) se induce experimentalmente en el encéfalo no lesionado por estímulos mecánicos, eléctricos o químicos y no provoca muerte celular. El córtex afectado por este fenómeno presenta una oxigenación tisular inicialmente normal y un flujo sanguíneo cerebral regional aumentado, seguido de una moderada hipoperfusión en la fase de repolarización8. La PID o CSD-like tiene unas características similares a la CSD clásica, pero se inicia de forma espontánea en las áreas de penumbra isquémica10.
En este trabajo pretendemos describir la metodología y los resultados preliminares del estudio de los fenómenos CSD y CSD-like en un grupo piloto de pacientes con TCE o infarto maligno de la arteria cerebral media (IMACM) estudiados mediante ECoG, definiendo la frecuencia y características de estos fenómenos.
Pacientes y métodosSe trata de un estudio observacional llevado a cabo por la Unidad de Investigación de Neurocirugía y Neurotraumatología del Hospital Universitario Vall d’Hebron (HUVH), con la participación multidisciplinar de los Servicios de Neurofisiología Clínica, Medicina Intensiva, Neurorrehabilitación y, en paralelo, con otras líneas de investigación de la Unidad de Neurovascular de este centro. Los pacientes incluidos en este estudio forman parte del estudio multicéntrico internacional CoOperative Study on Brain Injury Depolarisations (COSBID). El proyecto fue valorado y aceptado por el Comité de Ética del HUVH (PI080480).
Durante un periodo de 19 meses (02/2009–08/2010) se incluyeron 16 pacientes consecutivos que cumplieron los siguientes requisitos: A) Criterios de inclusión, 1) pacientes con un TCE en el que la tomografía computarizada cerebral objetivó lesiones intradurales tributarias de tratamiento quirúrgico o pacientes con un IMACM sometidos a craniectomía descompresiva; 2) edad entre 18 y 70 años; 3) consentimiento informado del familiar más cercano o representante legal del paciente aceptando las condiciones del estudio y sus potenciales riesgos. B) Criterios de exclusión, 1) pacientes en los que la monitorización invasiva estaba contraindicada (trastornos de la coagulación, infecciones, etc.); 2) pacientes con midriasis bilateral arreactiva u otros signos clínicos que sugirieran criterios de muerte cerebral.
Protocolo de tratamientoTodos los pacientes con un TCE incluidos fueron tratados de acuerdo con los protocolos asistenciales aprobados que ya se aplican en nuestra unidad y que siguen las pautas descritas en las guías de práctica clínica actualizadas en 2007 por la Brain Trauma Foundation11. El objetivo del tratamiento es mantener una presión intracraneal (PIC) inferior a los 20mmHg mediante medidas terapéuticas escalonadas. En los casos de hipertensión intracraneal refractaria, y cuando no existen lesiones ocupantes de espacio con un volumen significativo, los pacientes son tratados mediante craniectomía descompresiva.
Los pacientes con un IMACM fueron tratados mediante un protocolo consensuado que incluye básicamente la monitorización sistemática utilizada en los TCE graves, además de la práctica de una hemicraniectomía descompresiva en aquellos pacientes que presenten una desviación de la línea media ≥5mm y/o una PIC>20mmHg. De forma rutinaria no se administraron fármacos antiepilépticos profilácticos.
Para la sedación farmacológica se utilizó midazolam en aquellos casos con una PIC<20mmHg y/o que presentaron inestabilidad hemodinámica o hipotensión (tensión arterial sistólica<90mmHg). En los enfermos con PIC>20mmHg y estables hemodinámicamente la sedación alternativa fue propofol.
Todos estos pacientes requirieron monitorización invasiva sistemática con sensores de presión tisular de oxígeno (PtiO2) y microdiálisis cerebral de acuerdo con los protocolos asistenciales de nuestra unidad. Además también se efectuó una monitorización específicamente diseñada para este estudio mediante ECoG continua.
Monitorización sistemáticaTodos los pacientes incluidos en este estudio ingresaron en la Unidad de Cuidados Intensivos de Neurotraumatología del HUVH y fueron monitorizados sistemáticamente de acuerdo con las pautas asistenciales existentes, que incluyen siempre la monitorización de la presión arterial media y la colocación de un sensor de PIC, un catéter retrógrado en el bulbo de la yugular, un sensor de PtiO2 y un catéter de microdiálisis cerebral en el área de penumbra. La recogida de datos de los monitores se realizó mediante un sistema informatizado ya disponible en nuestra Unidad de Cuidados Intensivos que permite la integración simultánea de 16 señales analógicas (ML880 PowerLab 16/30, ADInstruments, New South Wales, Australia).
Monitorización específica para el estudioA los pacientes incluidos, además de la monitorización convencional, se procedió a colocar durante el acto quirúrgico una tira subdural de ECoG con una serie de 6 electrodos de platino de 5mm de diámetro, cuyos centros están situados a 10mm de distancia uno de otro (Wyler 6-electrode linear ECoG strip, AdTech, Racine, Wisconsin, EE. UU.) (fig. 1). Después de implantar los catéteres y los electrodos de ECoG, se practicó siempre una tomografía computadorizada de control para comprobar la correcta colocación de los sistemas de monitorización. Para minimizar los riesgos de infección, la monitorización se mantuvo un máximo de 7 días. La inserción de los electrodos de ECoG se efectuó una vez completada la evacuación de la lesión o la craniectomía descompresiva en los pacientes con un IMACM.
. Registro bipolar: canal A=1-2; canal B=2-3; canal C=3-4; canal D=4-5; canal E=5-6; canal F (adicional)=6-1.")
Después de completar la hemostasia, se colocó la tira de silicona con los electrodos en el córtex perilesional. El electrodo número 1 se colocó sobre córtex viable adyacente a la zona de resección, en el caso de contusiones, y en la zona periinfártica en los pacientes con IMACM. El cable se tunelizó subcutáneamente, exteriorizándolo de forma que pudiera retirarse sin necesidad de reabrir la herida una vez finalizada la monitorización. Se utilizó un electrodo de aguja de platino (Vyasis) como electrodo de referencia, que se insertó subcutáneamente a nivel mastoideo, y un electrodo de superficie autoadhesivo (Vyasis) como electrodo de tierra, colocado en región dorsal del hombro, ambos ipsilaterales a la craniectomía. Con este procedimiento se consiguió monitorizar aproximadamente 4cm de córtex perilesional.
Los electrodos de ECoG se conectaron al preamplificador (Octal Bioamplifier, ML138, ADInstruments, Australia) y este a su vez al integrador de señales. En cada registro se adquirieron 8 señales ECoG: 6 de los canales correspondían a registros bipolares (A=1-2, B=2-3, C=3-4, D=4-5, E=5-6 y F=6-1) (fig. 1) y 2 a registros referenciales (G=2-ref, H=5-ref), de acuerdo con el protocolo publicado por Fabricius et al.10 y validado por el COSBID.
En la pantalla de la estación de registro −PC con instalación del software LabChart (ADInstruments, Australia)− se visualizó la monitorización continua poligráfica correspondiente a los 6 canales bipolares en serie, la PtiO2, la presión arterial, la temperatura y la PIC.
Análisis electrocorticográficoPara el análisis ECoG se aplicó el protocolo de análisis publicado por Fabricius et al.10 y validado por el COSBID. Considerando las posibilidades iniciales de visualización en pantalla del programa LabChart de 16 canales en el momento en que se diseñó el estudio y que las condiciones técnicas en alguno de los canales no siempre fue óptima o se deterioró durante el transcurso de la monitorización, se seleccionan 4 de los registros bipolares de mayor calidad para llevar a cabo el análisis cuantificado ECoG.
La señal ECoG registrada en cada uno de los 4 canales seleccionados (A, B, C, D) fue tratada mediante 4 funciones matemáticas diferentes, obteniendo un total de 16 canales de análisis (fig. 2).
: canal A=electrodos 1-2; canal B=2-3; canal C=3-4; canal D=4-5; canal E=5-6; canal F=6-1. Se observan 2 episodios de CSD a las 5:32h y a las 6:22h, caracterizados por cambios esterotipados en la línea de base (SPC). Los números indican el electrodo que recoge la despolarización.")
Selección de 140min del registro ECoG de un paciente afecto de TCE. Registro bipolar (4 series de 6 canales): canal A=electrodos 1-2; canal B=2-3; canal C=3-4; canal D=4-5; canal E=5-6; canal F=6-1. Se observan 2 episodios de CSD a las 5:32h y a las 6:22h, caracterizados por cambios esterotipados en la línea de base (SPC). Los números indican el electrodo que recoge la despolarización.
Cada uno de los episodios de CSD identificables por la variación de la señal convencional EEG (canales 13-16) fueron realzados obteniendo la integración de la potencia (canales 9-12) y se correlacionaban con deflexiones lentas y estereotipadas (SPC, del inglés slow potential change) (canales 1-8). Se consideró la presencia de un episodio de CSD cuando se observó una reducción>50% de la potencia integrada durante más de 2min en al menos uno de los canales con un retraso de 0,5-20min entre electrodos, con posterior recuperación gradual de la actividad de fondo a la situación basal (fig. 2). Se consideró la presencia de un evento PID cuando se observaron al menos 2 episodios estereotipados de CSD sin haber alcanzado apenas la recuperación de la actividad de fondo. Se etiquetó el episodio de CSD/PID en caso de cumplir criterios mixtos de CSD o de PID en diversos canales.
En función de los criterios neurofisiológicos definidos por Fabricius et al.10 se cuantificó el número de CSD, de PID y de CSD/PID. Se hizo constar el día y hora en que tuvieron lugar estos fenómenos y se calculó la duración, canal por canal, de cada uno de los eventos.
En todos los pacientes se definieron los parámetros tiempo de despolarización mínima y tiempo de despolarización máxima. El tiempo de despolarización mínima se calculó sumando las duraciones menores para cada uno de los fenómenos CSD, es decir, la del canal de menor duración, y el tiempo de despolarización máxima se calculó sumando las duraciones de mayor valor para cada una de las CSD. También se calculó el índice de despolarización (cociente entre la duración total de las despolarizaciones y la duración total del registro).
Evaluación del resultado neurológicoEl resultado neurológico de los pacientes incluidos en el estudio se evaluó a los 6 meses (180±10 días) de la lesión, de acuerdo con la Extended Glasgow Outcome Scale12.
Análisis estadísticoEl análisis estadístico del estudio fue exclusivamente descriptivo. En todos los casos se evaluó la distribución normal o no-normal de las variables cuantitativas mediante el método de Shapiro-Wilks. Para el análisis estadístico, los días se expresaron en el horario de 24h. El tiempo transcurrido desde el inicio de los síntomas o desde el traumatismo hasta las 24h del mismo día se consideró para el análisis estadístico como día 0. En las variables continuas los estadísticos de resumen fueron la n, la media, la desviación estándar (DE), la mediana, el primer y tercer cuartil, y los valores mínimo y máximo.
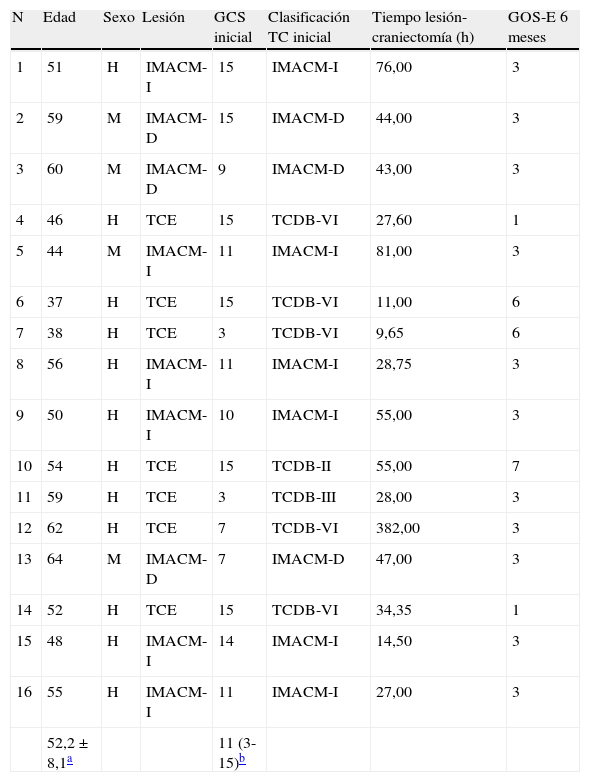
ResultadosEn este estudio se incluyeron 16 pacientes (12 hombres y 4 mujeres), 9 (56%) afectos de IMACM (6 pacientes con IMACM izquierdo y 3 con IMACM derecho) y 7 pacientes (44%) con un TCE moderado o grave (puntuación ≤13 puntos en la escala de coma de Glasgow), o leve, con un Glasgow inicial superior pero con un deterioro clínico y/o radiológico posterior que motivó un tratamiento quirúrgico (tabla 1). La edad media fue de 52,2+8 años, con un rango de 37-64 años. En 9 casos (56%) se observaron episodios de CSD y/o CSD-like (CSD/PID o PID), considerados como fenómenos SPC (tabla 2). Estos episodios se presentaron con mayor frecuencia en los pacientes con un IMACM (55%) que en los pacientes traumáticos (45%). El momento de presentación durante el transcurso de la monitorización fue muy variable, ya que en algunos pacientes la mayor parte de los fenómenos eléctricos se concentró en el primer día de registro, mientras que otros pacientes fueron presentando fenómenos SPC a lo largo de todo el periodo de monitorización ECoG (hasta el séptimo día). La duración y frecuencia de los mismos fue también muy variable (de 0 a 34 episodios).
Información clínica de los pacientes analizados
| N | Edad | Sexo | Lesión | GCS inicial | Clasificación TC inicial | Tiempo lesión-craniectomía (h) | GOS-E 6 meses |
| 1 | 51 | H | IMACM-I | 15 | IMACM-I | 76,00 | 3 |
| 2 | 59 | M | IMACM-D | 15 | IMACM-D | 44,00 | 3 |
| 3 | 60 | M | IMACM-D | 9 | IMACM-D | 43,00 | 3 |
| 4 | 46 | H | TCE | 15 | TCDB-VI | 27,60 | 1 |
| 5 | 44 | M | IMACM-I | 11 | IMACM-I | 81,00 | 3 |
| 6 | 37 | H | TCE | 15 | TCDB-VI | 11,00 | 6 |
| 7 | 38 | H | TCE | 3 | TCDB-VI | 9,65 | 6 |
| 8 | 56 | H | IMACM-I | 11 | IMACM-I | 28,75 | 3 |
| 9 | 50 | H | IMACM-I | 10 | IMACM-I | 55,00 | 3 |
| 10 | 54 | H | TCE | 15 | TCDB-II | 55,00 | 7 |
| 11 | 59 | H | TCE | 3 | TCDB-III | 28,00 | 3 |
| 12 | 62 | H | TCE | 7 | TCDB-VI | 382,00 | 3 |
| 13 | 64 | M | IMACM-D | 7 | IMACM-D | 47,00 | 3 |
| 14 | 52 | H | TCE | 15 | TCDB-VI | 34,35 | 1 |
| 15 | 48 | H | IMACM-I | 14 | IMACM-I | 14,50 | 3 |
| 16 | 55 | H | IMACM-I | 11 | IMACM-I | 27,00 | 3 |
| 52,2±8,1a | 11 (3-15)b |
D: derecha; GCS: escala de coma Glasgow; GOS-E: escala de resultados de Glasgow extendida; h: horas; H: hombre; I: izquierda; IMACM: infarto maligno de la arteria cerebral media; M: mujer; N: número de caso; TC: tomografía computarizada; TCDB: Traumatic Coma Data Bank; TCE: traumatismo craneoencefálico.
Clasificación en la TC inicial según la clasificación del TCDB: II, III, IV, V, VI.
GCS: puntuación en la escala de coma Glasgow (3-15).
GOS-E: escala de resultados de Glasgow extendida (1: muerte; 2: estado vegetativo; 3: discapacidad severa baja; 4: discapacidad severa alta; 5: discapacidad moderada baja; 6: discapacidad moderada alta; 7: buena recuperación baja; 8: buena recuperación alta).
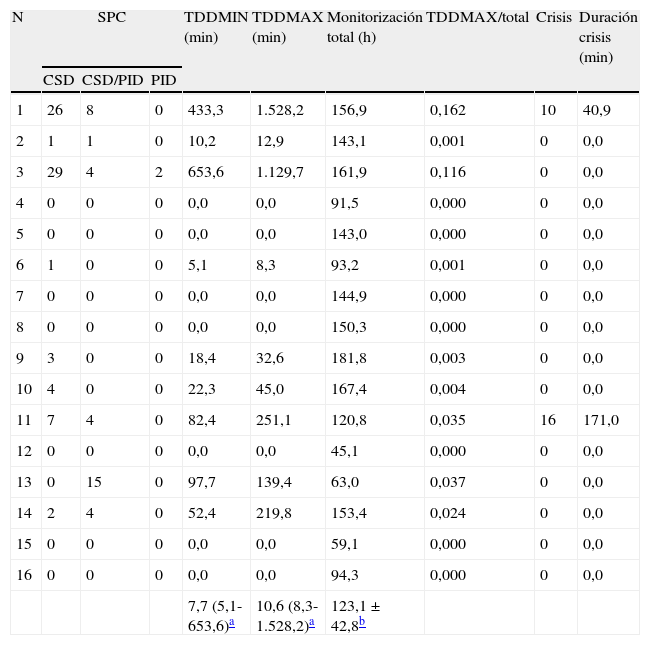
Análisis de los datos de electrocorticografía
| N | SPC | TDDMIN (min) | TDDMAX (min) | Monitorización total (h) | TDDMAX/total | Crisis | Duración crisis (min) | ||
| CSD | CSD/PID | PID | |||||||
| 1 | 26 | 8 | 0 | 433,3 | 1.528,2 | 156,9 | 0,162 | 10 | 40,9 |
| 2 | 1 | 1 | 0 | 10,2 | 12,9 | 143,1 | 0,001 | 0 | 0,0 |
| 3 | 29 | 4 | 2 | 653,6 | 1.129,7 | 161,9 | 0,116 | 0 | 0,0 |
| 4 | 0 | 0 | 0 | 0,0 | 0,0 | 91,5 | 0,000 | 0 | 0,0 |
| 5 | 0 | 0 | 0 | 0,0 | 0,0 | 143,0 | 0,000 | 0 | 0,0 |
| 6 | 1 | 0 | 0 | 5,1 | 8,3 | 93,2 | 0,001 | 0 | 0,0 |
| 7 | 0 | 0 | 0 | 0,0 | 0,0 | 144,9 | 0,000 | 0 | 0,0 |
| 8 | 0 | 0 | 0 | 0,0 | 0,0 | 150,3 | 0,000 | 0 | 0,0 |
| 9 | 3 | 0 | 0 | 18,4 | 32,6 | 181,8 | 0,003 | 0 | 0,0 |
| 10 | 4 | 0 | 0 | 22,3 | 45,0 | 167,4 | 0,004 | 0 | 0,0 |
| 11 | 7 | 4 | 0 | 82,4 | 251,1 | 120,8 | 0,035 | 16 | 171,0 |
| 12 | 0 | 0 | 0 | 0,0 | 0,0 | 45,1 | 0,000 | 0 | 0,0 |
| 13 | 0 | 15 | 0 | 97,7 | 139,4 | 63,0 | 0,037 | 0 | 0,0 |
| 14 | 2 | 4 | 0 | 52,4 | 219,8 | 153,4 | 0,024 | 0 | 0,0 |
| 15 | 0 | 0 | 0 | 0,0 | 0,0 | 59,1 | 0,000 | 0 | 0,0 |
| 16 | 0 | 0 | 0 | 0,0 | 0,0 | 94,3 | 0,000 | 0 | 0,0 |
| 7,7 (5,1-653,6)a | 10,6 (8,3-1.528,2)a | 123,1±42,8b | |||||||
CSD: despolarización cortical propagada (cortical spreading depression); h: horas; min: minutos; N: número de caso; PID: despolarizaciones periinfárticas (peri-infarct depolarizations); SPC: slow potential changes; TDDMAX: tiempo de despolarización máxima (total duration of depression–maximal); TDDMIN: tiempo de despolarización mínima (total duration of depression–minimal).
De los 9 pacientes con fenómenos SPC, solo 3 presentaron exclusivamente eventos CSD clásicos; 6 evidenciaron eventos CSD-like. Solo uno de estos 6 últimos pacientes presentó únicamente fenómenos CSD-like de tipo mixto CSD/PID, en ausencia de fenómenos CSD clásicos. La mayor parte de los fenómenos CSD-like se produjeron después de que se hubieran presentado fenómenos CSD. La tabla 2 muestra los resultados del análisis completo de ECoG de estos pacientes. El patrón EEG (ECoG) más frecuentemente observado fue el de salva-supresión. Dos de los 9 pacientes presentaron actividad epileptiforme ictal, constituida por actividad paroxística delta rítmica, en estrecha correlación temporal con los fenómenos CSD (fig. 3).
. Incremento fusiforme de la actividad bioeléctrica cerebral (canales B y C-EEG en escala 1k:1), correspondiente a actividad paroxística delta ictal (canales B, C y D en selección a escala 50:1) coincidiendo con una CSD. Cambio lento de potencial (SPC en canales 0,02Hz e Int) y depresión de la integral de la potencia (canales Int Pow).")
Selección del registro ECoG del paciente 1, afecto de IMACM-I. Registro bipolar (4 series de 4 canales). Incremento fusiforme de la actividad bioeléctrica cerebral (canales B y C-EEG en escala 1k:1), correspondiente a actividad paroxística delta ictal (canales B, C y D en selección a escala 50:1) coincidiendo con una CSD. Cambio lento de potencial (SPC en canales 0,02Hz e Int) y depresión de la integral de la potencia (canales Int Pow).
El paciente 1, afecto de un IMACM izquierdo, presentó el mayor número de episodios CSD y el mayor índice de despolarizaciones de todos los pacientes de la serie. Las despolarizaciones se detectaron entre el primer y el séptimo día de monitorización, con intervalos de tiempo y frecuencia muy variables. Este paciente presentó además 10 episodios de crisis comiciales subclínicas coincidiendo con los primeros 10 episodios de CSD que se produjeron durante las primeras 24h (fig. 3). Después de 26 episodios de CSD, aparecieron 8 eventos CSD-like. En el paciente 3, no obstante, la primera despolarización propagada cortical no se observó hasta haber transcurrido 76h desde la detección del IMACM. La mayoría de los episodios se presentaron durante el tercer día de evolución y continuaron produciéndose hasta una semana después del ictus.
El paciente 10 presentó un TCE grave con hemorragia subaracnoidea (HSA) y hematoma subdural izquierdo asociado a un foco de contusión cerebral, con una puntuación inicial de GCS de 15. En su evolución, este paciente requirió la resección de una contusión frontal izquierda al tercer día de ingreso. La totalidad de los eventos CSD4 se presentaron durante las 9 primeras horas de registro ECoG.
En el registro del paciente 11, afecto de un TCE grave, se cuantificó un total de 11 episodios (7 CSD clásica y 4 CSD-like), documentándose el inicio de actividad paroxística delta rítmica de carácter ictal 12min antes del último de los episodios de CSD-like.
El paciente 13 presentó un infarto maligno de arterias cerebral media y anterior derechas. Las despolarizaciones (15 en total), todas ellas de tipo mixto (CSD/PID), se presentaron de forma uniforme, al contrario de la presentación en cluster de otros pacientes.
El paciente 14 sufrió un TCE grave con hematoma subdural agudo izquierdo, asociado a una contusión frontal. Se practicó evacuación de la colección hemática y contusectomía frontal izquierda. El postoperatorio se complicó con la aparición de un síndrome de distrés respiratorio agudo. En este paciente se identificaron 6 fenómenos SPC (2 CSD y 4 CSD-like mixtos). Todos los fenómenos CSD-like tuvieron lugar después de los eventos CSD.
En 8 de los 9 pacientes que presentaron fenómenos CSD la PIC fue siempre inferior a 20mmHg. Respecto a la PtiO2, en uno de estos 9 pacientes el catéter de oximetría disfuncionó, en 6 casos la PtiO2 fue superior a 15mmHg y en solo 2 pacientes se objetivaron lecturas por debajo de los 15mmHg durante los episodios de despolarización.
Todos los pacientes incluidos en el estudio recibieron tratamiento con midazolam. Solo en 2 casos (un paciente con un TCE y otro con un IMACM) se administró propofol de forma adicional para complementar la sedación. De los 2 pacientes que recibieron propofol, uno (TCE) presentó un fenómeno CSD durante la administración del fármaco. Tres casos de IMACM fueron tratados con hipotermia. Todos ellos pertenecen al subgrupo de pacientes que presentaron fenómenos de despolarización.
A los 6 meses de la inclusión en el estudio, de los 16 pacientes, 11 eran todavía dependientes y 2 habían fallecido (tabla 1). Ninguno de los pacientes que presentaron fenómenos CSD-like eran funcionalmente independientes (puntuaciones Extended Glasgow Outcome Scale≤3 a los 6 meses de la lesión). En ninguno de los pacientes fallecidos la craniectomía descompresiva había sido demorada más de 48h desde su ingreso. La mortalidad a los 12 meses alcanzó el 31,25%.
DiscusiónNuestro estudio debe considerarse un estudio piloto en un grupo reducido de pacientes afectos de patología neurológica aguda (TCE o infarto maligno). La mitad de ellos presentaron durante su curso clínico episodios de CSD o CSD-like, lo que indica la elevada frecuencia de estas anomalías eléctricas en los pacientes con lesiones cerebrales agudas. Estos fenómenos pueden ocurrir sin que se identifiquen repercusiones en los parámetros monitorizados de forma rutinaria en estos pacientes. Con las limitaciones que nuestro estudio presenta en el tamaño muestral, la incidencia de estos fenómenos coincide con la detectada por otros autores. Diversos grupos han objetivado de forma inequívoca la existencia de fenómenos de CSD y CSD-like repetitivos en pacientes con un TCE, HSA e ictus isquémico13,14. Sin embargo, su incidencia no es universal, apareciendo con una frecuencia de entre el 50-60%, dependiendo del tipo de patología estudiada13,14.
La limitación más importante de todos estos estudios —incluido el nuestro— es que los pacientes estudiados han sido siempre intervenidos mediante una craneotomía, con lo cual la verdadera frecuencia de estos fenómenos es todavía incierta.
A nivel neuronal la CSD se caracteriza por una despolarización hasta un potencial de membrana de casi cero, lo que se logra mediante la combinación de un gradiente electroquímico reducido de potasio y la apertura de canales de sodio. Los fenómenos CSD-like aparecen espontáneamente en las áreas de penumbra isquémica en modelos de oclusión de la arteria cerebral media en babuinos, gatos y ratas. La ligadura de la arteria cerebral media en la rata demuestra de forma inequívoca que el incremento del volumen del infarto es una función lineal del número de PID15 y que la frecuencia de CSD-like es un factor independiente de la viabilidad final del tejido cerebral lesionado16. Los fenómenos PID no ocurren solo durante la oclusión vascular, sino también en la fase de reperfusión17. En modelos de isquemia, el tamaño final del infarto guarda una estrecha correlación con el número de fenómenos CSD-like detectados en la zona de penumbra16. Estos fenómenos son especialmente deletéreos para el tejido cerebral por la reducción del flujo sanguíneo cerebral regional en la región afectada. Este es el mecanismo más relevante en la expansión del core del infarto18. Esta hipoperfusión se asocia a la despolarización inicial y a la propagación de la onda generada16. En nuestra serie de pacientes, la presentación mayoritaria de los fenómenos CSD-like posterior en el tiempo a los fenómenos de CSD clásica durante el transcurso de la monitorización guardaría relación con el compromiso energético que sufre el tejido cerebral tras la cascada metabólica que suponen estos fenómenos eléctricos en el intento de restablecer el equilibrio iónico y, probablemente, con el reclutamiento de penumbra al core. Por otra parte, recientemente Nakamura et al.19 han demostrado en animales lisencefálicos que la propagación de las ondas de despolarización es repetitiva y suele hacerlo de forma circular alrededor de las lesiones isquémicas. La CSD-like se caracteriza por una depleción significativa de los niveles de glucosa en el tejido afectado. Parkin et al.20 demostraron este fenómeno en hematomas traumáticos. En pacientes con TCE existe una clara relación entre el número de ondas de despolarización y la depleción de glucosa. En estos pacientes la hipoglucemia tisular se asocia a un peor resultado neurológico20. En el ictus isquémico el término PID describe este mismo fenómeno en las áreas de penumbra isquémica, habiéndose demostrado que el número de PID deteriora el tejido estructuralmente intacto, favoreciendo su evolución a la necrosis.
Los fenómenos CSD/CSD-like cuando se inducen en un tejido cerebral sano, y con una perfusión normal, no inducen estrés metabólico persistente ni lesión celular. De hecho, la inducción de este fenómeno en animales sanos puede actuar incluso como un mecanismo neuroprotector de una forma similar a los fenómenos de precondicionamiento21,22. Sin embargo, la situación cambia cuando afectan a una región cerebral en la que existen situaciones de estrés metabólico o un tejido cerebral vulnerable (traumatismo, HSA o isquemia). En estos casos, estos fenómenos exacerban la lesión celular y aumentan el volumen final de tejido necrótico. Sus efectos deletéreos son debidos al enorme coste metabólico que supone para el encéfalo —con unas reservas energéticas y/o un flujo sanguíneo ya comprometidos— el restablecimiento del equilibrio iónico.
Takano et al.23 en un estudio de gran relevancia aportan evidencia experimental de que los fenómenos CSD y CSD-like contribuyen a expandir las lesiones secundarias en los TCE y en los ictus isquémicos. Estos autores demuestran que las ondas CSD clásicas, inducidas por potasio en animales, se asocian a una hipoxia tisular severa con depleción de NADH en el córtex por donde esta se propaga23. Esta hipoxia se acompaña con zonas parcheadas de incrementos del NADH sugestivos de hipermetabolismo23. Este estudio sugiere que incluso los fenómenos CSD clásicos producen importantes alteraciones metabólicas, de causa probablemente hipermetabólica por la activación de las bombas iónicas necesarias para restaurar la homeostasis del tejido cerebral afectado y repolarizar las neuronas.
Se ha demostrado que los fenómenos CSD-like inducen cambios en la expresión génica, estimulan la expresión de factores del crecimiento, de neurotransmisores excitadores y de mediadores de respuesta inflamatoria tales como la interleucina 1-β y el factor de necrosis tumoral α, y de metaloproteasas de matriz (MMP)9. Las MMP son una familia de proteasas neutras con gran capacidad lesiva cuando se sobreexpresan en lesiones del sistema nervioso central. La sobreexpresión de MMP, y en especial de las denominadas gelatinasas (MMP-2 y MMP-9), está implicada en diversos procesos tales como: 1) degradación de la membrana basal y disrupción de la barrera hematoencefálica, 2) invasión del tejido nervioso por células del torrente sanguíneo y 3) activación de citocinas y de sus receptores9. En un estudio reciente, nuestro grupo ha demostrado que la gelatinasa A se sobreexpresa a nivel sistémico y cerebral después de un TCE moderado o grave24. Estudios experimentales demuestran que los fenómenos CSD-like incrementan los niveles de MMP-99. Estos hallazgos coinciden con nuestras observaciones efectuadas en pacientes con TCE y microdiálisis de alta resolución24. Debido a su impacto sobre la barrera hematoencefálica, la sobreexpresión de MMP-9 puede facilitar el edema cerebral y el paso de moléculas del espacio intravascular al intersticio cerebral después de fenómenos persistentes de CSD-like.
A pesar de que estos fenómenos eléctricos no sean de naturaleza comicial, las crisis epilépticas, tal y como describen Fabricius et al.25, se presentan como un fenómeno satélite en relación con los episodios de CSD. De hecho, en uno de nuestros pacientes, el 29% de las despolarizaciones se asoció a la presencia de crisis y, en otro paciente el último de los eventos CSD-like fue precedido unos minutos de la presencia de crisis epilépticas, sugiriendo una posible implicación fisiopatológica de la actividad crítica como gatillo de los fenómenos de CSD.
En el TCE y en el accidente cerebrovascular isquémico masivo se produce una fase de deterioro diferido que a menudo se asocia a edema cerebral refractario al tratamiento y a un aumento significativo de la mortalidad y a un peor pronóstico funcional. La causa y los mecanismos implicados en este deterioro permanecen sin clarificar aunque los fenómenos CSD-like podrían contribuir a este deterioro. Hasta la fecha, los fenómenos de CSD-like han sido observados en pocos pacientes y su perfil metabólico ha sido poco estudiado en humanos. Nuestros datos preliminares confirman que estos fenómenos son frecuentes (56%), aunque no constantes, después de un TCE y un infarto maligno.
Los fenómenos de CSD-like contribuyen al deterioro metabólico del tejido cerebral, y no son simples epifenómenos en la lesión cerebral traumática ni en el ictus isquémico. La repercusión metabólica de estos fenómenos aumentaría el reclutamiento de las zonas de penumbra al tejido necrótico. Una mejor definición del perfil temporal de estos episodios y de los factores gatillo en series más grandes de pacientes podría definir una nueva diana terapéutica, ya que el bloqueo de las CSD y las CSD-like a nivel experimental se correlaciona de una forma clara con una mayor supervivencia del tejido cerebral dañado en todos los modelos de lesión cerebral aguda.
Financiación y agradecimientosEste estudio ha sido financiado en parte por el Fondo de Investigaciones Sanitarias de la Seguridad Social (FIS), número PI080480 y por la beca MAPFRE 05/2008 concedidas al Dr. J. Sahuquillo. Parte de los resultados de este estudio fueron publicados en la revista Trauma de la Fundación MAPFRE.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

