

INTRODUCCIÓN α
El pulmón supone una gran superficie de intercambio con el medio, y por lo tanto está equipado con una amplia serie de mecanismos de defensa frente a las diversas agresiones a las que se ve expuesto (patógenos, tóxicos, estímulos mecánicos, etc.)1. La lesión pulmonar aguda y el síndrome del distrés respiratorio agudo (SDRA) pueden producirse como consecuencia de alteraciones pulmonares y extrapulmonares y su fisiopatología es el resultado de una compleja interacción de mediadores humorales y células. De entre todos los mediadores (incluyendo derivados de ácido araquidónico, factores de coagulación, mecanismos de lesión y reparación de la membrana celular, etc.), citocinas y quimiocinas forman el grupo más importante, y nos centraremos en ellas. La apoptosis es otro mecanismo de lesión alveolar y endotelial que, por su estrecha relación con la respuesta inflamatoria, será tratado conjuntamente.
El pulmón en el SDRA es actualmente visto como un órgano más involucrado en el síndrome de disfunción orgánica múltiple como resultado del síndrome de respuesta inflamatoria sistémica, caracterizado por una activación de leucocitos mediada por citocinas. Los leucocitos en el pulmón, por lo tanto, responden y contribuyen al proceso inflamatorio en el SDRA. Existe una activación de leucocitos en la circulación sistémica que migran hacia la circulación pulmonar y posteriormente al intersticio, produciendo una respuesta inflamatoria local con posterior vertido de mediadores inflamatorios a la circulación general.
La patogénesis de la lesión pulmonar aguda se sustenta por tanto en 4 pilares fundamentales: a) daño endotelial y epitelial; b) activación de células inflamatorias; c) balance entre citocinas pro y antiinflamatorias y d) necrosis y apoptosis.
A la compleja interacción entre estos mecanismos se añade una quinta vía de lesión, producida por el estrés mecánico que supone la ventilación mecánica.
DAÑO ENDOTELIAL Y EPITELIAL
El endotelio tiene un papel clave en el desarrollo de la lesión pulmonar aguda. Se trata de un órgano implicado en numerosas funciones de regulación de la homeostasis. Muchas de sus funciones son constitutivas, mientras que otras son inducidas mediante la activación endotelial tras la exposición a estímulos proinflamatorios tales como las citocinas o endotoxinas2. Dentro de las primeras, destacan por su papel en la activación endotelial la interleucina (IL) 1 y el factor de necrosis tumoral α (TNF-α). Ambas son producidas por macrófagos activados, así como por las propias células endoteliales. Inducen un estado protrombótico e inflamatorio3, estimulando la producción de otras citocinas entre las que se incluyen quimiocinas, factores estimulantes de colonias, IL-6 y la propia IL-1, derivados del ácido araquidónico y óxido nítrico (NO), y aumentan la expresión de moléculas de adhesión3,4. Estudios realizados en seres humanos han demostrado en la microvasculatura pulmonar un aumento de expresión de los receptores tipo 2 de TNF (TNF-R2) y una mayor producción de IL-6 y IL-8 en pacientes con SDRA frente a controles, sugiriendo una mayor activación del endotelio pulmonar durante el desarrollo del SDRA o bien que ese endotelio es constitutivamente más reactivo en sujetos que posteriormente desarrollan SDRA5. El mecanismo último que subyace a la activación endotelial mediada por citocinas es la presencia de factores de transcripción (proteínas de unión al ADN que regulan la expresión de genes), entre los que destaca el factor nuclear αB (NF-αB), que se comentará más adelante.
La pérdida de la integridad epitelial en el SDRA tiene importancia tanto en el desarrollo como en la posterior recuperación del pulmón dañado. En condiciones normales, la barrera epitelial es mucho menos permeable que la endotelial6 y por tanto, el daño epitelial contribuye al edema alveolar. La lesión de los neumocitos tipo II (implicados en el transporte de iones, además de la producción de surfactante) altera el transporte normal de fluidos, impidiendo el aclaramiento del edema pulmonar y finalmente también se afecta la producción de surfactante. La alteración de la integridad epitelial resulta en la exposición de estructuras de la matriz extracelular, que interaccionan con macrófagos alveolares exacerbando la respuesta inflamatoria.
ACTIVACIÓN DE CÉLULAS INFLAMATORIAS
El estudio histológico del pulmón en pacientes con SDRA muestra un importante cúmulo de neutrófilos7, que parecen ser células clave en esta patología, sin estar demasiado claro si son causa o consecuencia del daño pulmonar agudo. También estarían implicados los macrófagos y eosinófilos, siendo responsables de la lesión pulmonar aguda en pacientes neutropénicos8-10. La interacción entre las células endoteliales y los leucocitos es un proceso fundamental en el desarrollo de SDRA, en tanto que constituye el primer paso en la migración de dichos leucocitos desde los capilares hacia el parénquima pulmonar y la subsiguiente respuesta inflamatoria. La fase inicial se caracteriza por la «captura y rodamiento» de los neutrófilos y está mediada por moléculas de adhesión celular de la familia de las selectinas11. La P-selectina se puede expresar en minutos en la superficie endotelial en respuesta a estímulos como la liberación de histamina, trombina, bradiquinina, leucotrieno C4 o radicales libres y a su vez interacciona con su receptor en los neutrófilos (P-selectina glucoproteína-1). La E-selectina es sintetizada por el endotelio tras la activación celular por citocinas como el TNF-α, IL-1 o la endotoxina. La segunda fase es la adhesión de los neutrófilos, que requiere la interacción entre la familia de las integrinas ß2 (concretamente CD11/CD18) expresadas en los neutrófilos con la molécula de adhesión intercelular (ICAM-1) expresada en las células endoteliales11. La expresión de esta última estaría estimulada por mediadores inflamatorios como el TNF-α, IL-1, interferón-α y endotoxina. Finalmente se produce una migración transendotelial de los neutrófilos a favor de un gradiente quimiotáctico y la molécula de adhesión PECAM-1 (platelet-EC adhesión molecule 1)11. Una vez en el espacio alveolar, los neutrófilos liberan radicales libres (que entre otras características alteran la barrera endotelial, aumentando su permeabilidad a través de la proteincinasa C), proteasas, leucotrienos y otras moléculas pro-inflamatorias como el factor activador de plaquetas. Los macrófagos alveolares también secretan citocinas, entre las que se incluyen la IL-1, 6, 8 y 10 y el TNF-α, que a su vez estimulan la quimiotaxis y activan a los neutrófilos.
Inicio de la respuesta inflamatoria
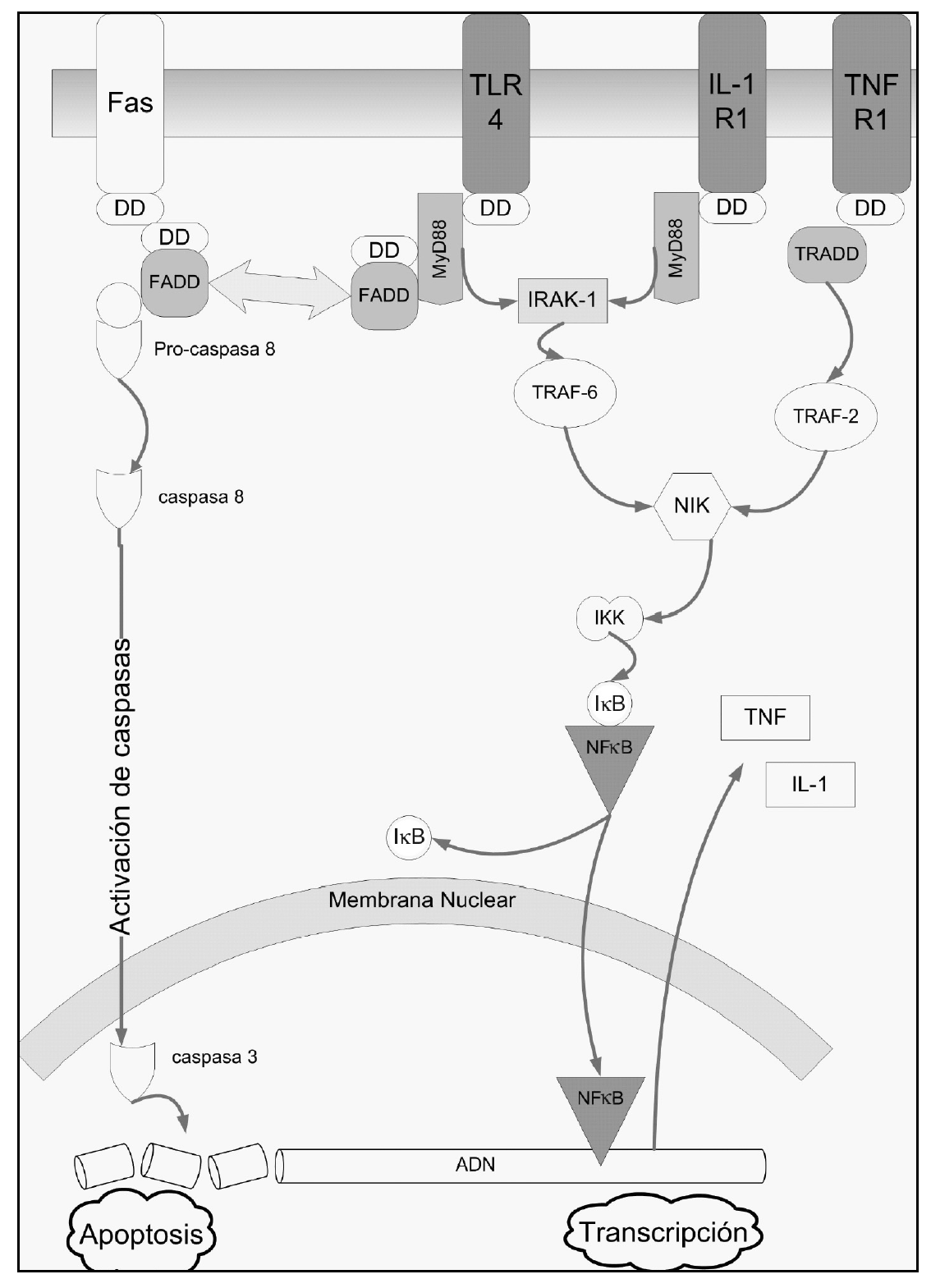
Las citocinas implicadas en la respuesta inflamatoria no se expresan constitutivamente en el pulmón, sino que se producen en respuesta a estímulos como por ejemplo la presencia de microorganismos patógenos. Aquí desempeñan un papel primordial los toll-like receptors (TLR) (fig. 1), que intervienen en el reconocimiento de dichos patógenos a través del reconocimiento de estructuras moleculares únicas presentes en ellos1. La transducción de señales está mediada por la molécula MyD88 que a través del reclutamiento de diferentes cinasas (IRAK, NIK) produce finalmente la liberación de NF-κB1. Uno de los primeros TLR caracterizados fue el TLR412 que reconoce el lipopolisacárido de bacterias gramnegativas a través de un mecanismo complejo en el que están implicadas diferentes proteínas que actúan como correceptores del TLR (CD14) o aumentando la respuesta al lipopolisacárido (MD-2). A su vez, el TLR2 reconoce componentes de bacterias grampositivas como el peptidoglicano13.
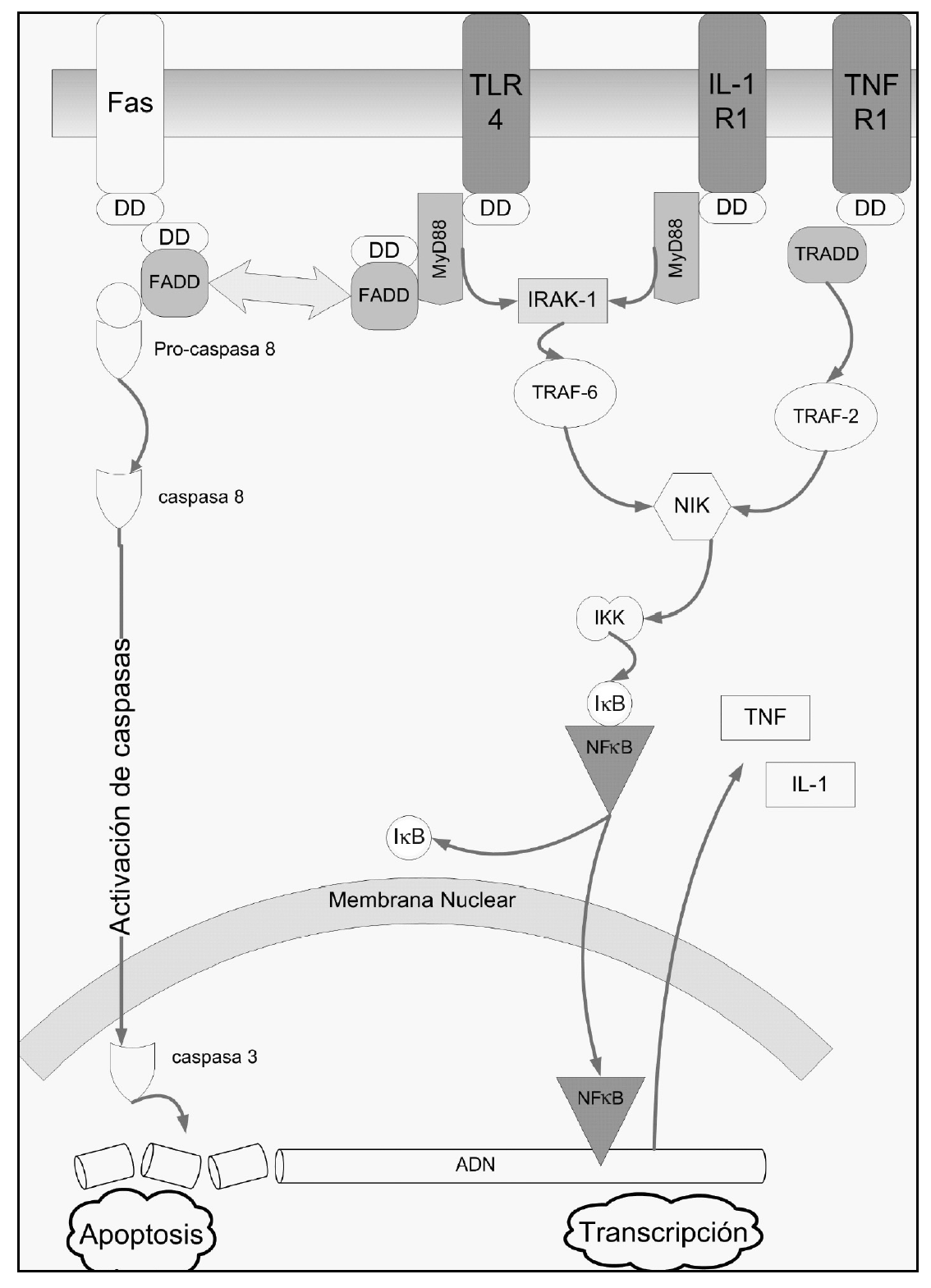
Figura 1. Representación de las vías de activación de la apoptosis (lado izquierdo de la figura, receptor Fas) y de varios mediadores inflamatorios. El TLR4 actúa como receptor del lipopolisacárido de bacterias gramnegativas. También se han representado las cascadas de la interleucina 1 (IL-1) y el factor de necrosis tumoral (TNF). Como puede verse, al final las vías de activación confluyen en la traslocación al núcleo celular del factor nuclear κB (NF-κB). Este factor activa la expresión de cientos de genes relacionados con la respuesta inflamatoria.También se han resaltado los death domains (DD) que comparten todos estos receptores. Estos dominios son la base estructural de la estrecha relación entre apoptosis y respuesta inflamatoria. TLR: toll-like receptors; DD: death domain; FADD: Fas-associed death domain.
El NF-κB es un factor de transcripción con estructura dimérica (habitualmente dos subunidades de 50 y 65 κDa), presente en el citoplasma celular. Allí se encuentra en una forma inactiva, unido al inhibidor del factor κB (IκB). Una gran variedad de señales (incluyendo el reconocimiento de antígenos en membrana, activación de receptores de citocinas, TLR, etc.) provoca la activación de las cinasas del IκB (IKK). La fosforilación del IαB provoca su separación del NF-κB. Este último se trasloca al núcleo celular, donde activa la expresión de cientos de genes, incluyendo genes de citocinas, moléculas de adhesión celular, receptores para la adhesión de neutrofilos, etc. Como puede adivinarse, gran parte de estos productos ejercen un feed-back positivo sobre la actividad del NF-κB. Esta es la base principal de la respuesta inflamatoria exacerbada que puede llevar al síndrome de disfunción multiorgánica.
El NF-κB parece ser el principal activador intracelular de la respuesta inflamatoria. Se ha encontrado una mayor activación de NF-κB en pacientes con lesión pulmonar aguda14, y se ha implicado también en la patogénesis de la lesión inducida por la ventilación mecánica15.
CITOCINAS
Un complejo grupo de citocinas y otros factores proinflamatorios inician y amplifican la respuesta inflamatoria en la lesión pulmonar aguda y el SDRA. Asimismo, en el alveolo se encuentran también inhibidores endógenos de estas citocinas proinflamatorias entre los que se incluyen el antagonista del receptor de IL-1 (IL-1ra), receptores solubles de TNF (sTNFR), autoanticuerpos contra la IL-8 y citocinas antiinflamatorias como la IL-10 y 1116. El balance entre estos mediadores pro y antiinflamatorios es determinante en el desarrollo de esta patología. Aunque la lista de mediadores implicada en la patogénesis de la lesión pulmonar aguda crece cada día, favorecida por el desarrollo de nuevas técnicas de biología molecular, describiremos solamente las características de las citocinas más claramente implicadas en este proceso.
IL-1ß (fig. 1)
Deriva predominantemente de macrófagos activados. Activa neutrófilos e induce aumento de expresión de moléculas de adhesión en leucocitos y células endoteliales17. Estimula la producción de múltiples citocinas y quimiocinas (IL-8, ENA-78, MCP-1, MIP-1α). Se ha detectado en el lavado broncoalveolar de pacientes con SDRA18,19, al igual que el IL-1ra, que antagoniza la unión de IL-1ß al receptor de superficie IL-1RI20,21. En voluntarios sanos se ha identificado una ratio IL-1ß: IL-1ra de 1, mientras que en pacientes con SDRA era de 10:120. Cuando se ha medido la actividad biológica de la IL-1 en pacientes con SDRA se ha encontrado una actividad neta proinflamatoria6,19,22.
TNF-α(fig. 1)
Al igual que el anterior, también deriva de macrófagos activados. Es una citocina proinflamatoria que estimula la producción de multitud de citocinas e interviene en la activación de neutrófilos23. Es uno de los principales mediadores del shock y se ha encontrado que la concentración de TNF-α y sus receptores (TNFR1 y TNFR2) está en relación directa con la gravedad de la enfermedad19,24.
IL-10
Es una citocina antiinflamatoria25 que inhibe la liberación de citocinas proinflamatorias como IL-1ß, IL-6 y TNF-α26,27. En un ensayo clínico con pacientes con SDRA de diferentes etiologías se demostró que los pacientes que tenían SDRA presentaban menores niveles de IL-10 circulantes y en lavado broncoalveolar (BAL) que aquellos con riesgo pero que no desarrollaban la enfermedad28. Esto subraya la importancia del imbalance proinflamatorio/antiinflamatorio en el SDRA, que a su vez podría ser reflejado mediante la ratio IL-10/TNF-α en el pulmón.
Factor inhibidor de la migración de macrófagos
Se trata de una citocina proinflamatoria29. Producida en el pulmón por macrófagos alveolares y células epiteliales bronquiales30,31. El factor inhibidor de la migración de macrófagos (MIF) y el TNF-α estimulan mutuamente su producción29. El MIF aumenta la producción de IL-8, potencia los efectos de la endotoxina y productos bacterianos de grampositivos32 y es un regulador esencial de la respuesta de los macrófagos a endotoxina a través de su efecto sobre la expresión del TLR-433. A través de la supresión de la apoptosis, el MIF estimula la supervivencia y función de los macrófagos34.
IL-6
Producida por una gran variedad de células, incluidos monocitos, macrófagos, células endoteliales, fibroblastos y células del músculo liso en respuesta al estímulo por endotoxinas, IL-1ß y TNF-α. Induce pirexia, activación y crecimiento de leucocitos, proliferación de células progenitoras mieloides y estimula la síntesis de proteínas de fase aguda. Los niveles de IL-6 circulantes han demostrado ser excelentes predictores de la gravedad del SDRA de diferentes etiologías35.
TGF-ß
Se trata de una citocina proinflamatoria que regula diversas actividades biológicas, incluyendo el crecimiento celular, apoptosis, diferenciación celular y síntesis de la matriz extracelular. Tiene un papel clave en el desarrollo de fibrosis pulmonar en la fase de regeneración tisular que sigue al SDRA36. En un estudio reciente se ha demostrado que la inhibición farmacológica del factor de crecimiento transformante ß (TGF-ß) protege a los ratones del edema pulmonar inducido por bleomicina o endotoxina de Escherichia coli23,37.
Factor estimulante de colonias de granulocitos y macrófagos
Producido por múltiples tipos celulares, está presente en el suero y la mayoría de los tejidos, aunque a bajas concentraciones. Interviene en la homeostasis de surfactante38. La deficiencia funcional del factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) conduce al desarrollo de proteinosis alveolar y defectos en los mecanismos de defensa pulmonar39. Niveles plasmáticos bajos se han asociado a un peor pronóstico en pacientes sépticos40.
Quimiocinas
Los neutrófilos son reclutados hacia el alveolo desde la circulación sistémica, gracias a factores quimiotácticos entre los que destacan las quimiocinas CXC41. Las quimiocinas son una familia de proteínas de pequeño tamaño (8-10 κDa) que se diferencian de las otras citocinas porque son las únicas que actúan sobre receptores acoplados a proteínas-G23. Son potentes factores quimioatrayentes de neutrófilos, eosinófilos, basófilos, monocitos, células dendríticas y linfocitos1. Se dividen en cuatro grupos en función de la disposición de residuos de cisteína en su extremo N-terminal: CXC (IL-8, GRO-α, GRO-ß, GRO-γ, ENA-78, GCP-2), CC (MCP-1, MIP-1α, RANTES), C y CX3C. A su vez, las CXC se pueden dividir en ELR positivas o ELR negativas basándose en la presencia o no de un dominio de tres aminoácidos (Glu-Leu-Arg: el motivo ELR). Las citocinas ELR + CXC son quimioatrayentes de neutrófilos y actúan como factores angiogénicos. Las ELR-CXC atraen monocitos. Las quimiocinas son producidas por un amplio grupo de células que incluyen monocitos, macrófagos alveolares, neutrófilos, plaquetas, eosinófilos, linfocitos, células epiteliales, fibroblastos, células del músculo liso, células mesoteliales y células endoteliales. Producen estas moléculas en respuesta a gran variedad de factores, entre los que se incluyen IL-1, TNF, complemento, leucotrienos e interferones1.
La IL-8, ENA-78 y GRO-α están presentes en concentraciones significativas en el BAL de pacientes con SDRA y su concentración se correlaciona con la concentración de polimorfonucleares20,42,43. La IL-8 es el principal quimioatrayente en el SDRA.
Hay dos receptores de CXC en los polimorfonucleares PMN: CXCR1 y CXCR2. El CXCR1 es el receptor fundamental implicado en la patogenia del SDRA, debido a su rápida cinética de expresión, internalización y reexpresión en la superficie celular44. Los ligandos más importantes de este receptor son IL-8 y GCP-2.
NECROSIS Y APOPTOSIS
La agresión (microbiológica, química o mecánica) a las células del epitelio alveolar puede desencadenar fenómenos de necrosis, caracterizada por un fallo global de todas las estructuras celulares, pérdida de la integridad de membrana y liberación del contenido celular. Todo esto desencadena una respuesta inflamatoria alrededor de la célula necrosada. Por el contrario, la apoptosis constituye un proceso fisiológico de reparación de tejidos que no implica fenómenos inflamatorios, y que ocurre de forma controlada, regulada por mediadores y cascadas enzimáticas intracelulares. La sobreexpresión o la inhibición de la apoptosis es un mecanismo de regulación de la remodelación tisular y, en el contexto de las células inflamatorias, de la respuesta inmune.
Mecanismo de apoptosis
El receptor Fas (CD95), presente en células alveolares epiteliales y que se incrementa en respuesta a estímulos inflamatorios, puede ser activado por su ligando (FasL) presente en la superficie de linfocitos citotóxicos. La unión de ligando y receptor provoca la activación de la procaspasa-8, que activa posteriormente a la caspasa-3. El proceso desencadena la fragmentación del ADN por endonucleasas. Otra vía de activación final de caspasa-3 se produce a través del citocromo C mitocondrial, por una vía dependiente de caspasa-945.
Papel de la apoptosis en la lesión pulmonar aguda
Los fenómenos de apoptosis participan en varios de los mecanismos fisiopatológicos implicados en la lesión pulmonar aguda:
1. Lesión del epitelio alveolar. La forma soluble de FasL (sFasL) es liberada por monocitos circulantes, pero no por macrófagos alveolares46. Se han encontrado niveles elevados de sFasL en procesos pulmonares como neumonías, bronquiolitis y SDRA47,48. La activación de la vía del Fas producida por la unión del ligando produce la apoptosis de las células del epitelio alveolar.
2. Regulación de la respuesta inmune. La apoptosis de los polimorfonucleares es un mecanismo de control de la respuesta inflamatoria. Varios mediadores (como el GM-CSF) aislados en el lavado broncoalveolar de pacientes con lesión pulmonar aguda son responsables de la inhibición de la apoptosis de PMN40,49. De esta manera se perpetúa la inflamación.
3. Extensión de la lesión a otros órganos. La liberación de sFasL a la circulación sistémica puede causar apoptosis en otros órganos. Se ha documentado la aparición de células apoptóticas en el riñón en animales con lesión pulmonar y ventilados con volúmenes elevados50.
Relaciones entre respuesta inflamatoria y apoptosis (fig. 1)
La apoptosis se ha considerado tradicionalmente un modo «silencioso» de muerte celular, en oposición a la necrosis (y así lo hemos dicho al inicio de esta revisión). Sin embargo, hay evidencia creciente de una estrecha relación entre los fenómenos de apoptosis y la respuesta inflamatoria.
Varios receptores de la membrana celular (como Fas, TNFR1, o la proteína adaptadora del TLR4 [MyD88]) comparten un mismo dominio en su porción citoplasmática, denominado dominio de muerte (death domain [DD]), que permite la interacción entre las proteínas que lo presentan. Se ha documentado que la estimulación de la vía del TNF desencadena tanto una respuesta proinflamatoria a través de la activación del NFκB como una vía proapoptótica51. De la misma manera, la activación del TLR4 por endotoxina activa ambos mecanismos. Para complicar más el cuadro, otros investigadores han documentado que la proteína FADD (Fas-associated death domain), adaptador intracelular del receptor Fas, es capaz de inhibir la activación de la vía del NFκB52.
A pesar de los resultados contradictorios, atribuidos a diferencias en los modelos utilizados, todos estos hallazgos sugieren que la activación de la respuesta inflamatoria y la apoptosis comparten una serie de mecanismos de regulación.
RESPUESTA INFLAMATORIA INDUCIDA POR LA VENTILACIÓN
Uno de los efectos de la ventilación mecánica, sobre todo con volúmenes altos y/o en pulmones con disminución de la compliancia, es la sobredistensión de los alveolos. Esto produce un reordenamiento del citoesqueleto y la membrana plasmática53, modifica la expresión de receptores de superficie, propaga señales a través de los canales intercelulares54, remodela la matriz55 e induce secreción de mediadores inflamatorios. Para todo ello, las células tienen mecanosensores que transducen el estímulo mecánico al interior de la célula y lo transforman en una respuesta química56. El estímulo mecánico se transduce en parte por la cascada de la MAPK (mitogen-activated protein kinase)57 y posiblemente la vías de c-fos58 y del factor NF-*B15. Estos factores de transcripción a su vez inducirían la expresión de genes codificadores de citocinas.
Aunque el efecto proinflamatorio de la ventilación mecánica sobre pulmones sanos es objeto de sonadas discusiones59,60, diversos estudios han demostrado que el empleo de volúmenes tidal muy elevados en pulmones sanos estimula la transcripción de genes y la secreción de mediadores inflamatorios58,61,62. Asimismo, existen estudios in vitro y en animales que demuestran un sinergismo entre la ventilación y diferentes estímulos, sugiriendo que ésta podría estimular a las células pulmonares a responder a dichos estímulos (bacterianos, químicos, etc.) con una hiperrespuesta inflamatoria. No obstante, no está claro si las fuerzas generadas durante la ventilación mecánica directamente estimulan las células pulmonares a inducir inflamación, o si la ventilación mecánica exacerba una inflamación ya existente en el pulmón.
El efecto de la ventilación mecánica sobre la respuesta inflamatoria se ha denominado biotrauma63. Los resultados obtenidos en grandes ensayos clínicos en los que se monitorizó una serie de citocinas en pacientes sometidos a volúmenes tidal bajos o elevados demostraron de manera sistemática un aumento de citocinas proinflamatorias en el grupo de volumen tidal elevado64. A pesar de los resultados a veces contradictorios de modelos experimentales65, es innegable que la ventilación mecánica puede modular la respuesta inflamatoria sistémica. Las estrategias ventilatorias más adecuadas, los mediadores más susceptibles de manipulación terapéutica y los grupos de pacientes que más se benefician son objetos de investigación actualmente.
CONCLUSIONES
Uno de los principales mecanismos de lesión pulmonar en el SDRA deriva de la activación del sistema inmune. La respuesta coordinada de mediadores y activación celular es necesaria para la eliminación de la causas desencadenantes y el inicio de los procesos de reparación. Pero la desregulación de esa respuesta lleva a un daño pulmonar grave. Los tratamientos encaminados a amortiguar la respuesta inmune conllevan el riesgo de entorpecer los mecanismos de defensa y cicatrización. Por eso es necesario un amplio conocimiento de los mecanismos de respuesta inflamatoria, que permita identificar a los pacientes que se beneficiarán de una estrategia terapéutica concreta.
Declaración de conflicto de intereses
Los autores han declarado no tener ningún conflicto de intereses
Financiado en parte por la beca FIS PI 04/1512.
*Éste es el tercero de 9 artículos de la Puesta al día en Medicina Intensiva: síndrome de distrés respiratorio agudo.
Correspondencia: Dr. G. Muñiz Albaiceta.
Servicio de Medicina Intensiva.
Hospital Universitario Central de Asturias.
C/ Celestino Villamil, s/n.
33006 Oviedo. España.
Correo electrónico: guillermo.muniz@sespa.princast.es
Manuscrito aceptado el 11-V-2006.

