




Traumatic brain injury (TBI) is a worldwide health problem that is especially prevalent in young adults. It is characterized by one or more primary injury foci, with secondary spread to initially not compromised areas via cascades of inflammatory response, excitotoxicity, energy failure conditions, and amplification of the original tissue injury by glia. In theory, such progression of injury should be amenable to management. However, all neuroprotective drug trials have failed, and specific treatments remain lacking. These negative results can be explained by a neuron centered approach, excluding the participation of other cell types and pathogenic mechanisms. To change this situation, it is necessary to secure a better understanding of the biological mechanisms determining damage progression or spread. We discuss the biological mechanisms involved in the progression of post-trauma tissue damage, including the general physiopathology of TBI and cellular mechanisms of secondary damage such as inflammation, apoptosis, cell tumefaction, excitotoxicity, and the role of glia in damage propagation. We highlight the role of glia in each cellular mechanism discussed. Therapeutic approaches related to the described mechanisms have been included. The discussion is completed with a working model showing the convergence of the main topics.
El traumatismo encéfalo craneano (TEC) es un problema de salud de distribución mundial, y es especialmente prevalente en la población adulta joven. Es característica la presencia de uno o más focos de daño, que luego progresan hacia áreas inicialmente no lesionadas, mediante cascadas de respuesta inflamatoria, excitotoxicidad, condiciones de falla energética, y la participación de la glía amplificando la respuesta tisular al daño inicial. Esta progresión es, en teoría, susceptible de una intervención terapéutica. Sin embargo, hasta ahora todos los estudios con fármacos neuroprotectores han fracasado, no existiendo un tratamiento específico efectivo. Los resultados negativos se explican en parte por el empleo de una estrategia centrada solo en las neuronas, sin considerar otras células participantes, u otros mecanismos patogénicos. Para cambiar este panorama, es necesario re-enfocar el problema a través de una mejor comprensión de los mecanismos que determinan la progresión del daño. En esta revisión discutiremos los principales mecanismos biológicos involucrados en la progresión del daño tisular post-trauma. Se aborda la fisiopatología general de los tipos de traumatismos, mecanismos celulares del daño secundario incluyendo inflamación, apoptosis, tumefacción celular, excitoxicidad, y participación de la glía en la propagación del daño. Se destaca el papel de la glía en cada uno de los mecanismos celulares mencionados. Se incluyen algunas aproximaciones terapéuticas relacionadas con los mecanismos descritos. Se finaliza con un diagrama general que resume los principales aspectos discutidos.
Traumatic brain injury (TBI) is the leading cause of morbidity–mortality in the population under 45 years of age. Each year, 1.4 million people suffer TBI in the United States1– one half of these cases being serious (Glasgow score<8)– and the mortality rate is ∼35% yearly. In Europe the situation is no different, with an estimated incidence of 235 cases per 100,000 inhabitants/year and a mortality rate of ∼11%.2 In addition, TBI is a known risk factor for the development of chronic neurodegenerative diseases such as Alzheimer's disease and Parkinson. Accordingly, because of its high incidence in the active or working population and the associated morbidity–mortality, TBI constitutes an important public health problem. However, no specific and effective treatments are available for preventing the associated damage.3 The present review discusses the main biological mechanisms involved in the progression of post-trauma damage. Improved understanding of the problem is a first step towards the development of effective management strategies. Table 1 shows the experimental studies commented in this review, which evaluate therapeutic approaches associated with the different biological mechanisms studied.
Summary of the experimental studies commented in this review that examine therapeutic approaches. The model of damage used is specified, along with the biological mechanism investigated and the main results obtained.
| Study | Model and species | Drug and therapeutic effect | Results |
| Clausen et al.22 | Controlled cortical impact, mice | IL-1β neutralizing antibody; (antiinflammatory) | Reduction of inflammatory response, brain loss and cognitive defect |
| Clark et al.27 | Controlled cortical impact, rats | Caspase-3 inhibitor tetrapeptide, (antiapoptosis) | Reduction of apoptosis and brain loss, without effect upon cognitive evaluation |
| Scheff et al.28 | Controlled cortical impact, rats and mice | Cyclosporine, (antiapoptosis) | Reduction of lesion volume |
| Mbye et al.33 | Controlled cortical impact, mice | NIM811 versus cyclosporine, (antiapoptosis) | Similar reduction of cytoskeletal fragmentation, neurodegeneration and neurological dysfunction |
| Kikuchi et al.43 | Unilateral hypoxia–ischemia, rat | Edaravone, (free radical binder) | Reduction of aquaporin 4 expression, infarct area and cognitive defect. |
| Frantseva et al.23 | Trauma in hippocampal organotypic slice culture, rats | Carbenoxolone, octanol and RNAi Cx43, (GJC block) | Reduction of cell death progression after trauma |
Depending on the biomechanics of the traumatism, TBI is classified as either focal or diffuse. Focal damage secondary to direct contact comprises epi- and subdural hematomas, contusions and intraparenchymal hematomas.4 In contrast, diffuse damage caused by acceleration/deceleration is characterized by a predominance of diffuse axon damage and brain edema.4
In the first hours following TBI, the brain presents islets of damaged cells in cases of focal injury, while the alterations are found to be more extensive in the case of diffuse injury. In both cases necrotized neurons and non-neuron cells are observed, with hemorrhagic foci that can conform intraparenchymal hematomas when sufficiently extensive.5 Variable degrees of edema and cellular swelling or tumefaction are also seen. Diffuse axon damage in turn is characterized by a broad and asymmetrical distribution of axon tumefaction phenomena.5
Spread of damageIn both focal and diffuse injuries, once direct tissue lesions have been established, a complex damage progression phenomenon develops, involving local and systemic factors that are globally known as secondary damage mechanisms (Fig. 1).
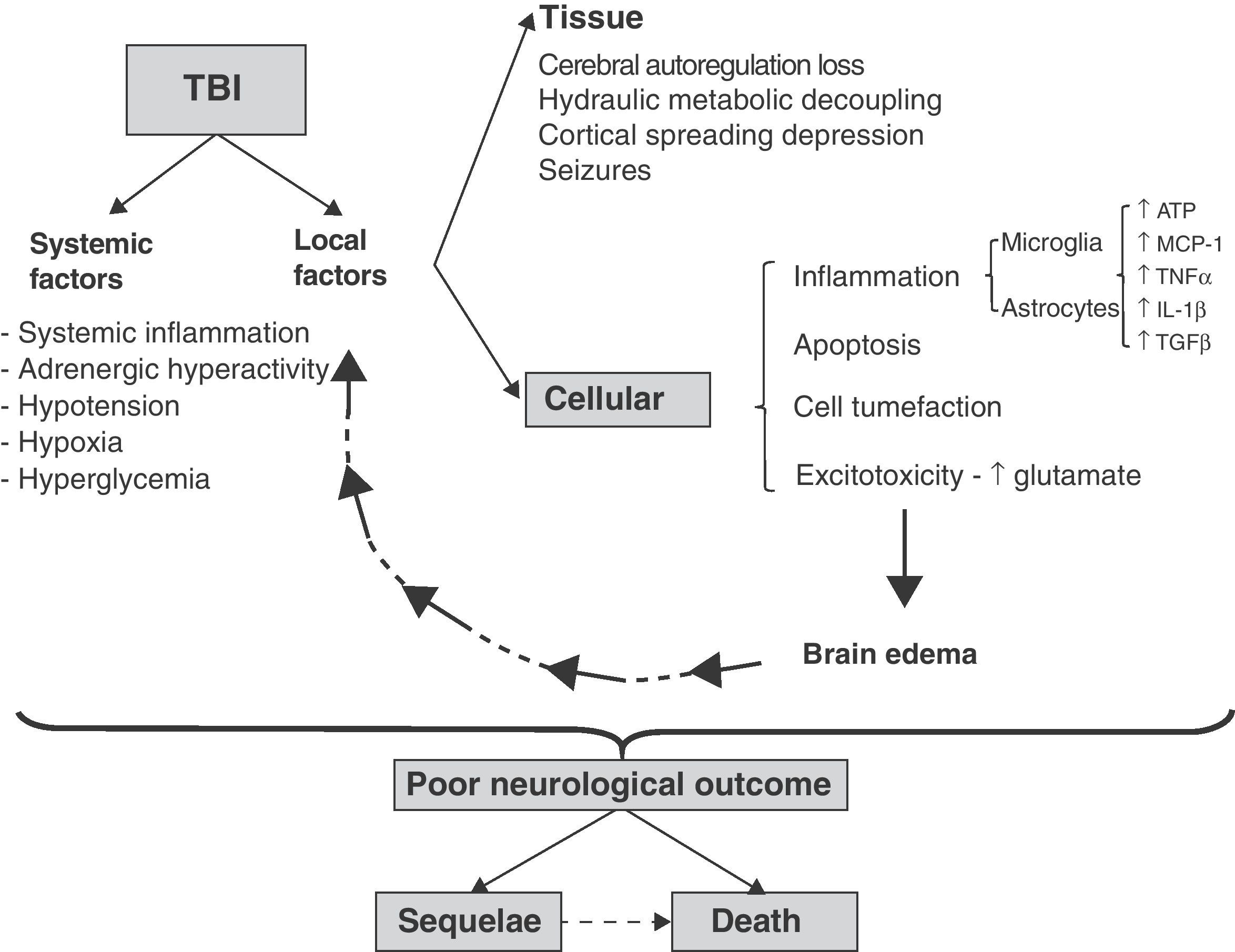
Cellular and molecular mechanisms involved in traumatic brain injury (TBI). Schematic representation of the inter-relationship between local and systemic factors in traumatic brain injury. Among the local factors, those referred to the cells appear to be particularly important for the amplification and progression of damage, causing it to become independent of the initial lesion. For further information on the tissue and systemic factors (see reference 4).
Among the systemic factors that worsen secondary damage, mention must be made of hypotension and hypoxia. Ninety percent of the patients that die as a result of severe TBI present findings compatible with ischemia; hypotension and hypoxia are observed in over 30% of the patients and represent two of the five most solid mortality predictors in TBI.6
The local elements of damage progression can be subdivided into tissue and cellular factors. Among others, the tissue factors comprise those dependent upon the relationship between brain perfusion pressure and brain blood flow.6 Following direct tissue damage, brain blood flow decreases, with the generation of physiopathological conditions similar to those of ischemia.4 In addition, extra-axial lesions are produced (contusions, hematomas, etc.) that cause damage secondary to compression and herniation of the brain parenchyma. In a later stage intracranial hypertension (ICH) can develop secondary to an increase in brain volume, due to the presence of extra-axial lesions and/or brain edema and the rigid nature of the skull dome.6 The cellular factors in turn correspond to pathogenic mechanisms that are common to both neurons and glial cells, such as the presence of inflammation, apoptosis, cellular edematization and excitotoxicity. The glia plays an intrinsic pathogenic role in the spread of damage, coordinating and amplifying each of the above-mentioned cellular pathogenic mechanisms. The systemic and local factors interact with each other and are reinforced as a result, causing damage progression to become independent of the initial cause of injury (Fig. 1) and the clinical presentation to be heterogeneous. Since the end result depends on both the initial and secondary damage, and the latter in turn interacts with local and systemic factors, there are many pathological differences among different patients, with an increase in the observed variability. This in turn greatly complicates the definition of therapeutic targets and the conduction of clinical trials.
One way to deal with such heterogeneity is to design “personalized” strategies based on the genotype of the individual patient. In this context, genetic association studies have demonstrated that certain genes present polymorphisms that are correlated to the post-TBI clinical outcome.7 In a recent metaanalysis of 2527 patients, isoform ApoE4 of apoprotein E was found to be associated with a poor clinical outcome 6 months after TBI.8 In contrast, a functional polymorphism of the p53 tumor suppression factor was associated with a poor prognosis at the time of discharge among TBI patients admitted to Intensive Care.9 The genes encoding for the interleukins are also interesting candidates for study. The repetition of two alleles of the interleukin-1 receptor antagonist (IL-1ra) gene was related to a more serious initial presentation but with a better ulterior clinical outcome.10
InflammationThe post-TBI inflammatory response is mediated by circulating leukocytes recruited towards the damaged zone, and by resident cells (microglia and astrocytes). The microglial cells are resident macrophages of the central nervous system (CNS), and are responsible for the innate immune response of the nervous system.11 Even in the absence of damage or exogenous stimulation, the microglial cells are involved in constant vigilance functions, with continuous remodeling of their activities. In this context, they are able to inspect the entire brain parenchyma within approximately 2h.12 Following stimulation, these cells are activated and take part in migration, phagocytosis and antigen presentation phenomena, and in the production of cytokines, free radicals and other soluble factors.11 Thus, the microglial cells function as damage sensors, coordinators and effectors of the inflammatory response of the CNS.
The blood–brain barrier (BBB) is often ruptured following TBI, with endothelial activation and the recruitment and infiltration of circulating leukocytes. In turn, the microglial cells are activated and migrate towards the site of damage13 through a series of mechanisms including monocyte chemoattractant protein-1 (MCP-1)14 and cytokines such as transforming growth factor β (TGF-β).15 Both the infiltrating leukocytes and the activated resident cells release proinflammatory cytokines, nitric oxide and free oxygen radicals.
Astrocytes also participate in the neuroinflammatory response. Those that survive the initial cellular damage undergo a phenotypic change known as reactive astrocytosis, characterized by hypertrophy, hyperplasia and an increased expression of the intermediate filament called glial fibrillary acidic protein (GFAP). This phenotypic change is mediated by tumor necrosis factor α (TNF-α) and interleukin 1-β (IL1-β).16 In moderate and focal trauma, astrocytosis contains the damaged area and favors neuroplastic remodeling (dendritic arborization and synaptic remodeling).17 However, in more severe TBI, astrocytosis can give rise to a glial scar that posteriorly complicates the neuroplastic changes required for post-injury functional accomodation.16
The different mediators commented above contribute to the perpetuation of neuroinflammation. However, this neuroinflammatory response can have either neuroprotective or neurotoxic effects, depending on the type and magnitude of the initial damage and on the context in which inflammatory response takes place.18
Inflammation appears to be directly correlated to the magnitude of TBI, and appears to be at least partially responsible for the development of ICH. In a clinical series of 23 patients with severe TBI, the levels of TNF-α, IL-1β, IL-1α, IL-6 and IL-8 in cerebrospinal fluid (CSF) measured with an ELISA technique were seen to increase early within the first 6h, reaching a maximum after 12h, and showing higher levels in those patients that developed ICH.19
On the other hand, the production of neurotrophic factors induced by cytokines exemplifies the potential neuroprotective role of inflammation. In a study of 22 patients with severe TBI, both IL-6 and nerve growth factor (NGF) were found to be increased in CSF. The elevation of NGF was detected simultaneously or following the appearance of IL-6. Posteriorly, the CSF of patients with TBI stimulated the production of NGF in astrocyte cultures−a situation not seen with the CSF of controls without TBI. This in vitro response in turn was inhibited using IL-6 neutralizing antibodies.20
In treatment terms, corticosteroids have not afforded benefits in terms of ICH control, despite their potent antiinflammatory action, and in fact there is evidence suggesting that these drugs can actually increase TBI mortality.21 However, there is a lack of clinical experience with more selective antiinflammatory management approaches such as the use of anti-cytokine antibodies, which could prove effective. In a murine model of focal TBI, a study was made of the intrathecal administration of an IL1-β neutralizing antibody. Its administration started 5min after trauma and was continued for 14 days. The neutralizing antibody was seen to reduce microglial activation, neutrophil infiltration, the loss of brain tissue and the neurological defects secondary to trauma.22
ApoptosisVariable degrees of brain parenchymal cell damage are observed in the context of TBI. Severe TBI results in rapid necrosis, with generation of the above-mentioned foci of direct cell damage. However, sublethal damage conditions, or the spread of damage signals to areas at a distance from the direct injury site, determine the occurrence of cell death through apoptosis in those regions.23
The existence of post-TBI apoptosis has been observed in both animal models and in patients.24–26 In rats, apoptosis is observed even after mild TBI.24 Apoptosis exhibits a particular space and time distribution, with maximum expression in the gray and white matter adjacent to the cortical impact site between 12 and 48h post-trauma, decreasing in proportion to the distance from the lesion, and proving detectable up to one week after injury. Later stages are characterized by the presence of apoptosis at a distance from the trauma site, in the ipsilateral thalamic region from 72h post-TBI, exhibiting a delayed time course with respect to the corresponding cortical zone.24
From the therapeutic perspective, Clark et al.27 reported that the intrathecal infusion of a caspase-3 (an apoptosis-executing enzyme) inhibiting tetrapeptide in a rat model of TBI reduces apoptosis and significantly lessens brain tissue loss as evaluated three weeks after TBI. However, the authors found no differences in the evolution of the neurological defects.27
The immune suppressor cyclosporine also reduces post-TBI brain volume loss in different in vivo models.28,29 This drug prevents opening of the mitochondrial permeability transition pores, which occurs as a result of Ca2+ overload in mitochondria observed during ischemia–reperfusion episodes and after TBI.30–32 The non-immune suppressor analog NIM811 proved equivalent to cyclosporine in reducing fragmentation of the cytoskeleton and in improving motor response in a model of TBI in mice.33 Considering the adverse effects of the immune suppressing activity of cyclosporine, NIM811 offers a comparatively better safety profile.
Cell tumefactionBrain edema and the associated complications are responsible for approximately 50% of all TBI deaths.34 In 1967, Klatzo used a model of cold-induced trauma to describe two types of brain edema, depending on its origin: vasogenic (extracellular) edema and cytotoxic edema, which more correctly corresponds to cell tumefaction or swelling.35 Recently, neuroimaging studies have found cytotoxic edema to be the prevalent component of brain edema following focal and diffuse TBI.36
The astrocyte is the main type of cell suffering post-traumatic tumefaction.37 Considering that astrocytes are 10 times more numerous than neurons,38 they are seen to be the main type of cell affected by cytotoxic edema. Although many factors have been implicated, the mechanisms underlying tumefaction are still little understood. It has been postulated that the above described direct cell damage and initial cerebral hypoperfusion give rise to the consumption of ATP, with energy failure and alteration of the transmembrane ion gradients. This in turn produces neuron depolarization and the release of glutamate, which induces an increased influx of Na+ and Ca2+ into the cell. The resulting Na+ overload cannot be compensated by the Na+/K+ ATPase pump system, due in part to the concomitant energy failure−thus favoring the penetration of water into the cell and an increase in cell volume.39
The aquaporins (Acp) are membrane channels that represent the main route for the passage of water through the cell membranes. In the brain, the main aquaporin is Acp-4, and its distribution is strongly polarized in the ependymal cells and astrocytes.40 Using a model of cytotoxic brain edema in knock-out (KO) mice, a significant reduction in edema was observed, as well as improvement of the neurological parameters in mice lacking Acp-4.41 In the same KO-Acp-4 mice, a reduction in brain water content and improved survival were observed in a model of vasogenic edema secondary to water intoxication.41
Two recent studies have explored the pharmacological manipulation of Acp-4. Using liposome-reconstituted Acp-4, it was seen that Zn2+ blocks water passage quickly and reversibly.42 In turn, Kikuchi et al.,43 using edaravone (a free radical binder) as treatment in a model of reversible focal brain ischemia, recorded a decrease in the expression of Acp-4 ipsilateral to the infarction 24h after reperfusion, accompanied by a reduction in the size of the infarction zone and in the neurological defect.43
ExcitotoxicityWithin minutes after TBI, experimental and clinical studies have found the extracellular glutamate levels to rise sharply.44,45 This increase in extracellular glutamate is mainly a consequence of massive neuron depolarization due to the traumatism and the associated energy failure commented above. This excess of glutamate in turn induces increased Na+ and Ca2+ influx to the cell, and the resulting intracellular Ca2+ overload triggers cell damage mechanisms that finally lead to apoptosis through caspase activation.46,47 Glutamate excitotoxicity initially takes place in the neurons, under conditions in which the astrocytes represent the main defense by reuptaking glutamate. The astrocytes are able to take up glutamate thanks to the presence of the glutamate glutamate-transporter 1 (GLT-1) and the glutamate-aspartate transporter (GLAST). The cells then metabolize the glutamate to glutamine and buffer it spatially in combination with other astrocytes coupled through gap junction channels (GJCs).46 The GJCs are formed by the junction of two hemi-channels (HCs), composed of connexin hexamers (Cxs).48 However, in a second stage, if high extracellular glutamate levels persist, the astrocytes actually enhance excitotoxicity, since the expression of glutamate transporters decreases49 and the intracellular Na+ overload can invert transport,50 favoring glutamate release from the astrocyte and thereby increasing its extracellular concentrations even further, with the result of enhanced excitotoxic damage.
Since the neurotoxic effects of glutamate have been known for over 50 years,51 it was initially thought that excitotoxicity could be the main physiopathological factor in TBI. However, this idea has been reconsidered in the light of the many negative results obtained in therapeutic studies in this field.52 The use of N-methyl-d-aspartate (NMDA) type glutamate receptor antagonists such as selfotel, or of endogenous blockers of the receptor such as magnesium sulfate, has failed to yield benefits in clinical studies of TBI patients.53,54 Moreover, both selfotel and magnesium sulfate increased mortality among the patients compared with placebo.53,54
Spread of damage, role of the gliaThe glial cells, and the astrocytes in particular, originally regarded as simply neuron supporting elements, received little attention in the study of TBI. However, in the last decade the role of these cells has been reinterpreted. The astrocytes form the microarchitecture of the nervous system, express receptors and channels, and are structurally organized into networks that communicate through GJCs, establishing functional syncytia.55
The progression of cell damage could be facilitated by intercellular communication mediated through channels formed by Cxs. In response to stimulation with different neurotransmitters or external signals, the astrocytes can transmit specific messages to neighboring cells, fundamentally through calcium waves–a phenomenon now known as gliotransmission,56 in which the GJCs take part.57 In addition to the GJCs, there are free HCs in regions of the membrane that do not establish cell contacts−those communicating the interior of the cell with the extracellular medium. Through these hemi-channels the cells release biologically active molecules such as glutamate, ATP and prostaglandins.58 In this context, two calcium wave spreading pathways have been demonstrated: (a) an intracellular pathway mediated by GJCs that allows the passage of molecules such as inositol triphosphate (IP3), which induce Ca2+ release from intracellular reservoirs; and (b) an extracellular pathway mediated by HCs, where the main signaling molecule is ATP,48 which is freed into the extracellular medium, followed by signaling in distant cells through the P2 purinergic receptors.
It has been suggested that the GJCs allow the intercellular transmission of apoptotic signals,59 involving the mediation of one of the neighboring damage mechanisms –a phenomenon that has also been described in models of damage produced by the transection of cell culture monolayers.60 The Cxs are classified according to their molecular weights (in kilodaltons, e.g.: Cx30, Cx43, Cx46).61 Recently, it has been shown that HCs formed by Cx43 contribute to the spread of apoptosis in cells distant from the initial stimulus.62
The modulation of HC aperture is associated with the development of damage. In this context, a delay in HC aperture induced via metabolic inhibition is correlated to a delay in cell damage63 and to a decrease in ATP release.64 One of the proposed triggering factors of HC aperture has been an increase in extracellular potassium (Ke+).65 Interestingly, one of the phenomena characteristic of trauma is the release of intracellular contents, including K+, into the interstice, as a result of direct cellular damage−reaching Ke+ concentrations of over 60meq/l in vivo.66 This Ke+, by inducing Ca2+ channel opening67 or HC aperture,68 generates an intracellular Ca2+ overload. The latter in turn can induce both necrosis and apoptosis through caspase activation.47
From the therapeutic perspective, the blockade of connexin conformed channels (GJCs and HCs) could have a beneficial effect in TBI. In a model of neurotrauma in hippocampal organotypic slice cultures, the use of GJC blockers, carbenoxolone and octanol, was seen to limit the area of cell damage to only a single point of impact, preventing its spread to the rest of the slice. In addition, the reduction of Cx43 through the use of RNAi also reduced the spread of post-trauma cell death.23
Diffuse axon damageSpecial mention should be made of diffuse axon damage, where the lesions fundamentally affect the white matter. The latter is composed of axon bundles with sheathes of myelin produced by the oligodendrocytes (OLGs), with the added presence of astrocytes and endothelial cells. Cell-to-cell interaction between the OLGs and the neuron axons is critical for the formation of myelin and for white matter maintenance and repair.69
Diffuse axon damage is found in up to 41% of all patients with TBI,70 and is responsible for variable degrees of persistently altered consciousness. Clinically, diffuse axon damage is characterized by rapid progression towards coma, with no specific intracranial lesion identifiable from the neuroimaging studies. The histopathological findings comprise axon swelling of diffuse and asymmetrical distribution, appearing a few hours after trauma. Concomitantly, hemorrhagic foci are seen along the affected axons, with the presence of amyloid peptide β (Aβ) precursor. Posteriorly axon degeneration is observed, with the development of retraction balls.5 These are produced when axon transport is interrupted as a result of cytoskeletal alterations−giving rise to swelling or tumefaction at the site of disconnection (hence the term “retraction ball”).71
Diffuse axon damage has a clinical outcome different to that of focal damage, with the triggering of a cascade of changes leading to secondary axon disconnection. According to Farkas and Povlishock72, there are two forms of axon damage. A first form is characterized by increased axon permeability, mitochondrial swelling and degradation of the cytoskeleton, with compaction of the neurofilaments. The second form is characterized by arrested axon transport and the development of axonal swelling, but without any increase in axon permeability.72 Unfortunately, from the therapeutic perspective, diffuse axon damage is the area least studied to date, and few studies have been able to offer advances in terms of treatment perspectives.
ConclusionsTraumatic brain injury is characterized by the presence of foci of direct cell damage. However, different local as well as systemic mechanisms cause the progression of damage to become independent of the initial lesion. The local mechanisms include particularly the glial cells, both microglia acting as damage sensors and coordinating the inflammatory response, with the secretion of many cytotoxicity mediators, and highly inter-connected astrocytes that actively participate in the inflammation process, the production of free radicals, cell tumefaction, excitotoxicity and the spread of apoptosis. The HCs and GJCs in turn are able to amplify several of these responses by either facilitating the spread of damage, or contributing directly to the latter. On the other hand, the OLGs play a preponderant role in white matter homeostasis−this being the main tissue affected in diffuse axon damage. Most clinical studies have been designed with the aim of activating neuroprotective mechanisms. However, given the negative results obtained, and considering the physiopathological mechanisms underlying the spread of post-TBI damage, a new approach seems needed in order to address the mechanisms of spread of damage mediated by the glia.
Financial supportProject PMD-10/09, Research Board of the Faculty of Medicine-PUC (M.R.).
Doctoral Thesis aid 2010, AT-24100202 CONICYT (M.R.).
Fondecyt 1070591 (J.C.S.); Fondecyt 1090353 (R.vB.).
Conflict of interestThe authors declare no conflicts of interest.
Please cite this article as: Rovegno M, et al. Mecanismos biológicos involucrados en la propagación del daño en el traumatismo encéfalo craneano. Med Intensiva. 2012;36:37–44.

