





Resuscitation for patient with sepsis and septic shock
More infoLa sepsis y el shock séptico son el resultado de una respuesta inadecuada del huésped a una infección, lo que produce disfunción orgánica. La progresión de esta afección se manifiesta por la aparición de estadios clínicos sucesivos, derivados de la respuesta inflamatoria sistémica secundaria a la activación de diferentes mediadores inflamatorios que conducen a la disfunción orgánica. Hay gran cantidad de evidencia sobre el papel de la endotoxina en la patogénesis de la sepsis y el papel fundamental que tiene en el inicio de la respuesta inflamatoria en la sepsis causada por microorganismos Gram-negativos. La activación de la cascada de coagulación en los pacientes con sepsis es parte de la respuesta inmunitaria adaptativa del huésped a la infección. El endotelio es el blanco principal en la sepsis, el cual es metabólicamente activo y puede responder a diversos estímulos fisiológicos y patológicos (activación endotelial). Adaptar los tratamientos a pacientes específicos es una estrategia obligada y personalizada para centrarse en la coherencia hemodinámica. La investigación futura debe responder a si las estrategias dirigidas a reclutar microcirculación pueden mejorar los desenlaces clínicos de los pacientes.
Sepsis and septic shock result from an inadequate host response to an infection, which causes organ dysfunction. The progression of this condition is manifested by the occurrence of successive clinical stages, resulting from the systemic inflammatory response secondary to the activation of different inflammatory mediators, leading to organ dysfunction. There is a high burden of evidence on the role of endotoxin in the pathogenesis of sepsis and its crucial role in triggering the inflammatory response in sepsis caused by gram-negative bacteria. The coagulation cascade activation in sepsis patients is part of the host's adaptive immune response to infection. The endothelium is the main target in sepsis, which is metabolically active and can
La sepsis y el shock séptico son el resultado de la respuesta inadecuada del huésped a una infección que ocasiona disfunción de uno o más órganos1. La respuesta inflamatoria en la mayoría de los individuos es adaptativa y contribuye a controlar la infección. Sin embargo, en la sepsis se produce un desequilibrio entre los mecanismos proinflamatorios y antiinflamatorios. La progresión de esta condición se caracteriza por la aparición sucesiva de estadios clínicos que son el resultado de la respuesta inflamatoria sistémica secundaria a la activación de diferentes mediadores inflamatorios que conducen a disfunción orgánica2 (Fig. 1). Esto incluye diferentes procesos fisiológicos, como la activación de diferentes líneas celulares (monocitos, macrófagos, neutrófilos, células endoteliales, plaquetas), la producción local y sistémica de citocinas, la estimulación de la cascada de proteínas plasmáticas (como el sistema del complemento), la activación de las vías de coagulación intrínseca (sistema de contacto) y extrínseca y el sistema fibrinolítico, la producción de mediadores lipídicos y la activación de la vía del óxido nítrico (NO), la producción de radicales libres, la estimulación de los linfocitos B y T y sus productos y fenómenos de proteolisis3, entre otros4.

Evolución de la sepsis. Con la interacción de PAMP y DAMP con la célula presentadora de antígenos, se inicia la cascada inflamatoria de la inmunidad innata y adaptativa. La inflamación causa daño tisular y secundariamente disfunción orgánica. En algunos pacientes se genera un estado de inmunoparálisis caracterizado por la aparición de infecciones nosocomiales y oportunistas y reactivaciones virales debido a la apoptosis del tejido inmunitario y la incapacidad relativa de las células T. DAMP: damage- associated molecular patterns; PAMP: pathogen-derived molecular patterns; TLR: toll-like receptor.
La respuesta inflamatoria frente a una infección se inicia mediante el reconocimiento de moléculas derivadas del patógeno (pathogen-derived molecular patterns [PAMPS]) y derivadas del huésped (damage-associated molecular pat- terns [DAMP]). El reconocimiento de estas moléculas por receptores específicos (toll-like receptors [TLR]) en la superficie de las células presentadoras de antígenos (CPA) inicia la cascada de transcripción de sustancias que producen inflamación, aumento del metabolismo celular y activación de la inmunidad adaptativa5. Funcionan como CPA las células del sistema monocito-macrófago, las células dendríticas, los linfocitos B y cualquier célula del organismo que exprese en su membrana determinantes antigénicos relacionados con proteínas del complejo mayor de histocompatibilidad. Cuando la inflamación es excesiva, causa daño tisular y secundariamente disfunción orgánica. Asimismo, en algunos pacientes la inflamación excesiva se acompaña posteriormente de un estado de inmunoparálisis caracterizado por la aparición de infecciones nosocomiales, infecciones oportunistas y reactivaciones virales debido a la apoptosis del tejido inmunitario e incapacidad relativa de las células T6,7. La respuesta inmunológica disminuida frente a determinadas infecciones8 se debe a fenómenos como la metilación del ADN en los monocitos.
Uno de los inductores de la sepsis mejor conocidos es la endotoxina o lipopolisacárido (LPS) de las bacterias Gram- negativas9. Hay múltiples pruebas de la implicación de la endotoxina en la patogénesis de la sepsis. La administración de LPS a animales de experimentación e individuos sanos reproduce la mayor parte de las alteraciones hemodinámicas derivadas del shock séptico y la disfunción multiorgánica. En el sistema cardiovascular, incrementa la frecuencia cardiaca y el índice cardiaco y disminuye las resistencias vasculares sistémicas, lo que produce hipotensión. En la microvasculatura, activa la coagulación, la hemostasia primaria, el complemento y el sistema fibrinolítico. El LPS actúa en el endotelio vascular inhibiendo la acción de anticoagulantes endógenos, induciendo la síntesis de radicales libres e incrementando la síntesis de NO.
Además, la endotoxina activa los macrófagos induciendo la síntesis de citocinas y los neutrófilos causando lesión endotelial mediante la producción de aniones superóxido y enzimas proteolíticas10. El sistema del monocito-macrófago es la principal diana de la acción del LPS. El suero humano y las membranas celulares contienen proteínas y receptores que se unen de forma específica al LPS para regular la compleja respuesta inmunológica del huésped a esta toxina bacteriana. Además, se han caracterizado factores solubles que se unen a la endotoxina y modulan sus efectos biológicos, como la proteína de unión al LPS (LPS-binding protein [LBP]) y la proteína bactericida incrementadora de la permeabilidad (bacterici- dal permeability increasing protein [BPI]). El CD14, receptor del LPS, tiene el papel de transferir la señal del LPS vía TLR-
También tiene interés y aplicabilidad la presepsina, o segmento N-terminal del CD14, que se utiliza como biomarcador de la sepsis11,12. Asimismo, durante la hemoadsorción de endotoxina en los pacientes con sepsis13, se han descrito extensos cambios en la modulación de la respuesta inflamatoria.
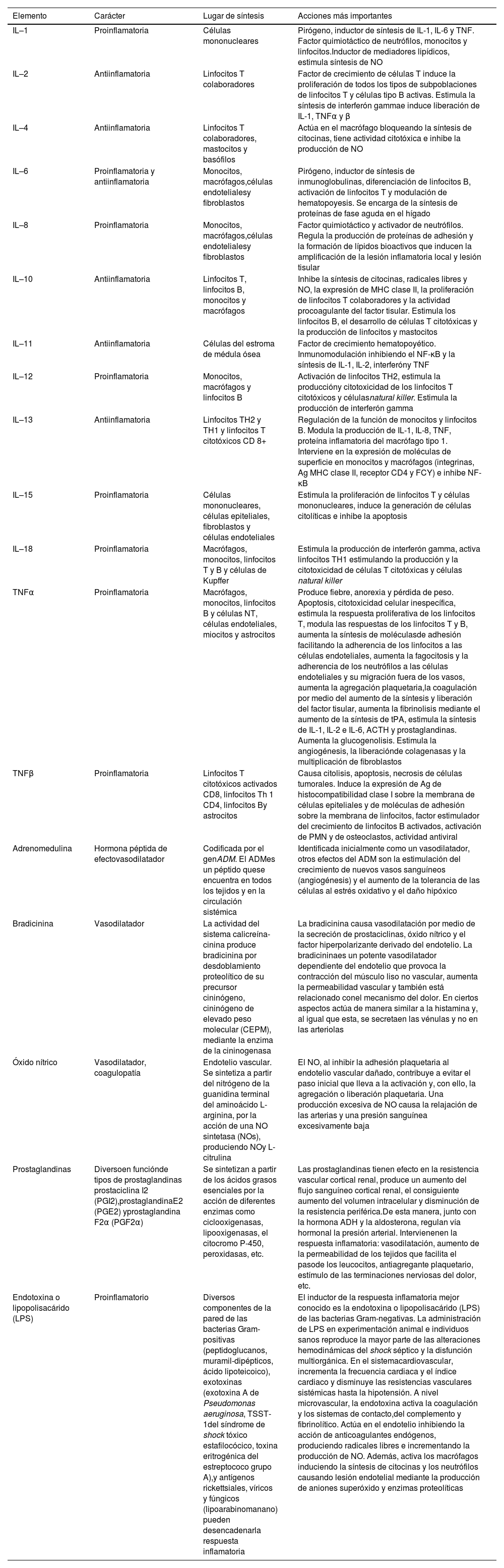
La unión de PAMP y DAMP a las CPA induce una activación celular que lleva a la translocación nuclear del factor NF-kB en las células B activadas y, en consecuencia, la expresión de citocinas proinflamatorias (interleucina [IL] 1, IL-2, IL- 18), factor de necrosis tumoral alfa [TNFα] e interferón [IFN]). De forma secundaria, se produce la activación de otras citocinas (IFNy, IL-6, IL-8), del sistema de complemento y de la cascada de coagulación, así como la regulación a la baja de los componentes del sistema inmunitario adaptativo14. En la Tabla 1 se mencionan los efectos de los principales mediadores inflamatorios en la sepsis.
Princ ipales mediadores infl amatorios en la sepsis
| Elemento | Carácter | Lugar de síntesis | Acciones más importantes |
|---|---|---|---|
| IL–1 | Proinflamatoria | Células mononucleares | Pirógeno, inductor de síntesis de IL-1, IL-6 y TNF. Factor quimiotáctico de neutrófilos, monocitos y linfocitos.Inductor de mediadores lipídicos, estimula síntesis de NO |
| IL–2 | Antiinflamatoria | Linfocitos T colaboradores | Factor de crecimiento de células T induce la proliferación de todos los tipos de subpoblaciones de linfocitos T y células tipo B activas. Estimula la síntesis de interferón gammae induce liberación de IL-1, TNFα y β |
| IL–4 | Antiinflamatoria | Linfocitos T colaboradores, mastocitos y basófilos | Actúa en el macrófago bloqueando la síntesis de citocinas, tiene actividad citotóxica e inhibe la producción de NO |
| IL–6 | Proinflamatoria y antiinflamatoria | Monocitos, macrófagos,células endotelialesy fibroblastos | Pirógeno, inductor de síntesis de inmunoglobulinas, diferenciación de linfocitos B, activación de linfocitos T y modulación de hematopoyesis. Se encarga de la síntesis de proteínas de fase aguda en el hígado |
| IL–8 | Proinflamatoria | Monocitos, macrófagos,células endotelialesy fibroblastos | Factor quimiotáctico y activador de neutrófilos. Regula la producción de proteínas de adhesión y la formación de lípidos bioactivos que inducen la amplificación de la lesión inflamatoria local y lesión tisular |
| IL–10 | Antiinflamatoria | Linfocitos T, linfocitos B, monocitos y macrófagos | Inhibe la síntesis de citocinas, radicales libres y NO, la expresión de MHC clase II, la proliferación de linfocitos T colaboradores y la actividad procoagulante del factor tisular. Estimula los linfocitos B, el desarrollo de células T citotóxicas y la producción de linfocitos y mastocitos |
| IL–11 | Antiinflamatoria | Células del estroma de médula ósea | Factor de crecimiento hematopoyético. Inmunomodulación inhibiendo el NF-κB y la síntesis de IL-1, IL-2, interferóny TNF |
| IL–12 | Proinflamatoria | Monocitos, macrófagos y linfocitos B | Activación de linfocitos TH2, estimula la produccióny citotoxicidad de los linfocitos T citotóxicos y célulasnatural killer. Estimula la producción de interferón gamma |
| IL–13 | Antiinflamatoria | Linfocitos TH2 y TH1 y linfocitos T citotóxicos CD 8+ | Regulación de la función de monocitos y linfocitos B. Modula la producción de IL-1, IL-8, TNF, proteína inflamatoria del macrófago tipo 1. Interviene en la expresión de moléculas de superficie en monocitos y macrófagos (integrinas, Ag MHC clase II, receptor CD4 y FCY) e inhibe NF-κB |
| IL–15 | Proinflamatoria | Células mononucleares, células epiteliales, fibroblastos y células endoteliales | Estimula la proliferación de linfocitos T y células mononucleares, induce la generación de células citolíticas e inhibe la apoptosis |
| IL–18 | Proinflamatoria | Macrófagos, monocitos, linfocitos T y B y células de Kupffer | Estimula la producción de interferón gamma, activa linfocitos TH1 estimulando la producción y la citotoxicidad de células T citotóxicas y células natural killer |
| TNFα | Proinflamatoria | Macrófagos, monocitos, linfocitos B y células NT, células endoteliales, miocitos y astrocitos | Produce fiebre, anorexia y pérdida de peso. Apoptosis, citotoxicidad celular inespecífica, estimula la respuesta proliferativa de los linfocitos T, modula las respuestas de los linfocitos T y B, aumenta la síntesis de moléculasde adhesión facilitando la adherencia de los linfocitos a las células endoteliales, aumenta la fagocitosis y la adherencia de los neutrófilos a las células endoteliales y su migración fuera de los vasos, aumenta la agregación plaquetaria,la coagulación por medio del aumento de la síntesis y liberación del factor tisular, aumenta la fibrinolisis mediante el aumento de la síntesis de tPA, estimula la síntesis de IL-1, IL-2 e IL-6, ACTH y prostaglandinas. Aumenta la glucogenolisis. Estimula la angiogénesis, la liberaciónde colagenasas y la multiplicación de fibroblastos |
| TNFβ | Proinflamatoria | Linfocitos T citotóxicos activados CD8, linfocitos Th 1 CD4, linfocitos By astrocitos | Causa citolisis, apoptosis, necrosis de células tumorales. Induce la expresión de Ag de histocompatibilidad clase I sobre la membrana de células epiteliales y de moléculas de adhesión sobre la membrana de linfocitos, factor estimulador del crecimiento de linfocitos B activados, activación de PMN y de osteoclastos, actividad antiviral |
| Adrenomedulina | Hormona péptida de efectovasodilatador | Codificada por el genADM. El ADMes un péptido quese encuentra en todos los tejidos y en la circulación sistémica | Identificada inicialmente como un vasodilatador, otros efectos del ADM son la estimulación del crecimiento de nuevos vasos sanguíneos (angiogénesis) y el aumento de la tolerancia de las células al estrés oxidativo y el daño hipóxico |
| Bradicinina | Vasodilatador | La actividad del sistema calicreína-cinina produce bradicinina por desdoblamiento proteolítico de su precursor cininógeno, cininógeno de elevado peso molecular (CEPM), mediante la enzima de la cininogenasa | La bradicinina causa vasodilatación por medio de la secreción de prostaciclinas, óxido nítrico y el factor hiperpolarizante derivado del endotelio. La bradicininaes un potente vasodilatador dependiente del endotelio que provoca la contracción del músculo liso no vascular, aumenta la permeabilidad vascular y también está relacionado conel mecanismo del dolor. En ciertos aspectos actúa de manera similar a la histamina y, al igual que esta, se secretaen las vénulas y no en las arteriolas |
| Óxido nítrico | Vasodilatador, coagulopatía | Endotelio vascular. Se sintetiza a partir del nitrógeno de la guanidina terminal del aminoácido L-arginina, por la acción de una NO sintetasa (NOs), produciendo NOy L-citrulina | El NO, al inhibir la adhesión plaquetaria al endotelio vascular dañado, contribuye a evitar el paso inicial que lleva a la activación y, con ello, la agregación o liberación plaquetaria. Una producción excesiva de NO causa la relajación de las arterias y una presión sanguínea excesivamente baja |
| Prostaglandinas | Diversoen funciónde tipos de prostaglandinas prostaciclina I2 (PGI2),prostaglandinaE2 (PGE2) yprostaglandina F2α (PGF2α) | Se sintetizan a partir de los ácidos grasos esenciales por la acción de diferentes enzimas como ciclooxigenasas, lipooxigenasas, el citocromo P-450, peroxidasas, etc. | Las prostaglandinas tienen efecto en la resistencia vascular cortical renal, produce un aumento del flujo sanguíneo cortical renal, el consiguiente aumento del volumen intracelular y disminución de la resistencia periférica.De esta manera, junto con la hormona ADH y la aldosterona, regulan vía hormonal la presión arterial. Intervienenen la respuesta inflamatoria: vasodilatación, aumento de la permeabilidad de los tejidos que facilita el pasode los leucocitos, antiagregante plaquetario, estímulo de las terminaciones nerviosas del dolor, etc. |
| Endotoxina o lipopolisacárido (LPS) | Proinflamatorio | Diversos componentes de la pared de las bacterias Gram-positivas (peptidoglucanos, muramil-dipépticos, ácido lipoteicoico), exotoxinas (exotoxina A de Pseudomonas aeruginosa, TSST-1del síndrome de shock tóxico estafilocócico, toxina eritrogénica del estreptococo grupo A),y antígenos rickettsiales, víricos y fúngicos (lipoarabinomanano) pueden desencadenarla respuesta inflamatoria | El inductor de la respuesta inflamatoria mejor conocido es la endotoxina o lipopolisacárido (LPS) de las bacterias Gram-negativas. La administración de LPS en experimentación animal e individuos sanos reproduce la mayor parte de las alteraciones hemodinámicas del shock séptico y la disfunción multiorgánica. En el sistemacardiovascular, incrementa la frecuencia cardiaca y el índice cardiaco y disminuye las resistencias vasculares sistémicas hasta la hipotensión. A nivel microvascular, la endotoxina activa la coagulación y los sistemas de contacto,del complemento y fibrinolítico. Actúa en el endotelio inhibiendo la acción de anticoagulantes endógenos, produciendo radicales libres e incrementando la producción de NO. Además, activa los macrófagos induciendo la síntesis de citocinas y los neutrófilos causando lesión endotelial mediante la producción de aniones superóxido y enzimas proteolíticas |
Los neutrófilos, además, se liberan en sus formas inmaduras desde la medula ósea. Al activarse por los mediadores, presentan capacidad fagocítica reducida15,16. Los neutrófilos activados formarán redes extracelulares en el tejido infla mado17, que inmovilizan al patógeno y estimulan otras sustancias inflamatorias. Estos procesos conllevan la activación de la cascada de la coagulación18 (Fig. 2). De hecho, los pacientes sépticos con una evolución clínica desfavorable presentan un aumento de las redes de neutrófilo tisular19.

Fisiopatología de la sepsis. La unión de PAMP y DAMP a las CPA induce una activación de elementos inflamatorios que conlleva la producción de citocinas, la activación de neutrófilos, la activación del sistema del complemento y de la cascada de coagulación y causará disfunción orgánica en caso de una respuesta desequilibrada. CID: coagulación intravascular diseminada; CPA: célula presentadora de antígenos; IL: interleucina; DAMPs: damage-associated molecular patterns; G-CSF/GM-CSF: factor estimulador de colonias; LPB: LPS-binding protein; PAMP: pathogen-derived molecular patterns; ROS: especies reactivas de oxígeno, TNF: factor necrosis tumoral; TLR: toll-like receptors.
El sistema del complemento también está implicado en la fisiopatología de la sepsis. Tanto la fracción 5a (C5a) como su receptor (C5aR) son actores relevantes. Tras la unión con los neutrófilos, este elemento del sistema de complemento penetra en la célula, donde PAMP y DAMP inducen la red neutrofílica que genera daño oxidativo y coagulopatía caracterizada por un estado protrombótico y bloqueo fibrinolítico20. Una activación excesiva de C5a causa apoptosis de los linfocitos y disfunción de los neutrófilos21.
Existe una heterogeneidad o variabilidad interindividual considerable en la respuesta inflamatoria descrita, que se modula por varios factores, como la hipogammaglobulinemia22, lo que motiva dificultades en el diagnóstico y el tratamiento23. Bermejo-Martin et al. han descrito hasta 10 inmunofenotipos distintos asociados con la inmunosupresión y el desequilibrio inmunológico en la sepsis. Los fenotipos que describen son las alteraciones cuantitativas y cualitativas en las CPA, los linfocitos B y T, las células NK y los neutrófilos. También describen alteraciones en la proporción de las células T reguladoras, el incremento en la expresión de la proteína programmed cell death protein-1 (PD-1), hipogammaglobulinemia, hipercitocinemia, disfunción del sistema de complemento y el importante papel de las redes extracelulares de neutrófilo en el desarrollo de la infección24. Los autores proponen que su estudio conduzca a un tratamiento personalizado. Este último aspecto no se trata en esta revisión.
3Endotelio y coagulopatía en la sepsisLa activación de la cascada de la coagulación en el paciente séptico se reconoce como parte de una respuesta inmunitaria adaptativa del huésped a la infección25. Sin embargo, la coagulación intravascular diseminada (CID), que puede aparecer en pacientes con sepsis, es el resultado de una activación desmedida y deletérea de la coagulación26. Se trata un síndrome adquirido caracterizado por activación de los factores de la coagulación originados a raíz de un daño en el lecho microvascular que puede llegar a causar disfunción orgánica dependiendo de su magnitud27. Alcanza una gran variedad de entidades clínicas, con fenómenos hemorrágicos en etapas iniciales28, alteraciones en las pruebas de coagulación o isquemia distal de las extremidades por fenómenos trombóticos29. Son 3 los factores clave que contribuyen en la CID: la activación de la cascada de la coagulación, la agregación plaquetaria y el daño endotelial.
4Activación de la cascada de coagulación y?bloqueo?fibrinolíticoLa sepsis se caracteriza por un estado protrombótico junto con bloqueo fibrinolítico, y ambos son los mecanismos fundamentales de la CID. Hay una serie de mediadores inflamatorios que promueven la activación de la trombina en la vía común de la coagulación como parte de la respuesta del huésped a la infección30. El factor tisular (FT), por su parte, como activador de la vía extrínseca, se expresa en macrófagos, monocitos y células endoteliales31. La acción conjugada de PAMP y DAMP es el factor determinante para el inicio del proceso. Además intervienen otros mediadores humorales, como IL-1, IL-6, TNFα, elastasa, catepsina G y el sistema del complemento, que también son facilitadores del proceso32. Incluso las redes extracelulares de neutrófilos activados también están implicadas33.
No se produce exclusivamente una activación de la cascada de la coagulación, sino de los elementos que la regulan, como el sistema fibrinolítico, que se alteran y entran en disfunción. Se hallan valores elevados del inhibidor tisular de plasminógeno, que producen un viraje a un estado de trombofilia en la CID34. De hecho, su concentración plasmática se ha relacionado con disfunción orgánica y mortalidad en la sepsis35. También hay una regulación a la baja de los inhibidores de la trombina36 y el plasminógeno37 e incremento del inhibidor del activador del plasminógeno, lo que causa un bloqueo fibrinolítico. También se observa reducción de las concentraciones plasmáticas de proteína C, por lo que se ha convertido en factor pronóstico de mortalidad38.
La inhibición de las proteínas procoagulantes por el endotelio se produce a través de los sistemas anticoagulantes fisiológicos: la vía de la proteína C, el sistema de la antitrombina III y el inhibidor del FT. En la sepsis se produce una disminución de estos inhibidores por consumo, reducción de su síntesis o degradación mediante elastasas. La consecuencia de ello es la aparición de un estado protrombótico caracterizado por una coagulación activada e insuficiencia de los sistemas inhibidores de la coagulación.
5Disfunción endotelialEl endotelio es el órgano diana fundamental de la sepsis39 y se caracteriza por ser metabólicamente activo y tener la capacidad para responder a diversos estímulos fisiológicos y patológicos. Entre sus funciones están el mantenimiento de la hemostasia y la fluidez de la sangre. El endotelio desempeña un papel clave en la regulación de la presión arterial y es fundamental en los mecanismos inflamatorios. Las células endoteliales mantienen un delicado equilibrio del tono vascular, la adhesión de las células sanguíneas y la coagulación. Por diversos mecanismos, el endotelio es capaz de controlar el tono vasomotor, mantener la fluidez de la san gre e intervenir en la respuesta inflamatoria. En condiciones fisiológicas, se caracteriza fundamentalmente por sus propiedades antitrombóticas, anticoagulantes, profibrinolíticas y antiagregantes plaquetarias. Como consecuencia de la estimulación de varias citocinas (TNFα, IL-1 y otros mediadores inflamatorios, como el complemento activado), estas propiedades endoteliales se ven profundamente alteradas y se produce una transformación endotelial denominada “activación del endotelio”. Esta se caracteriza por una superficie endotelial procoagulante, un bloqueo fibrinolítico mediado por liberación masiva del inhibidor del activador del plasminógeno, la expresión de moléculas de adhesión y la producción de mediadores inflamatorios y agentes vasoactivos40,41.
6Agregación plaquetariaLa trombocitopenia en la sepsis se debe a diversos factores. Fundamentalmente, los mediadores inflamatorios suprimen la producción plaquetaria. Otro mecanismo es el aumento del consumo, debido a que las plaquetas participan en la CID al ser activadas por la trombina, el sistema de complemento y el factor de von Willebrand (FvW)42.
7Alteraciones hemodinámicas macrovasculares y microvasculares en la sepsis y el shock séptico7.1Alteraciones macrocirculatorias en la sepsisA nivel macrohemodinámico, se ha considerado históricamente que el shock séptico ocurre en diferentes fases. En la primera fase hay una hipovolemia causada principalmente por pérdidas relativas de volumen, debidas al aumento de la capacitancia venosa y la disminución del volumen estresado que determina el retorno venoso43. También influyen en la hipovolemia las pérdidas absolutas de volumen debidas a fiebre, disminución de la ingesta, sangrado, pérdidas gastrointestinales, etc. La segunda fase es la hiperdinámica de la sepsis y la tercera, disfunción cardiaca que conduce a disfunción multiorgánica44, aunque la cardiomiopatía asociada a sepsis se puede adelantar y estar presente desde el inicio45.
Estas fases pueden ser intercambiables, siempre caracterizadas por ser un problema principalmente de carácter distributivo. La fase temprana hiperdinámica se caracteriza por alto gasto cardiaco, bajas resistencias periféricas e hipovolemia relativa. Después hay una fase hipodinámica con bajo gasto, pobre perfusión distal y disfunción multiorgá- nica46.
Clásicamente, la evaluación macrohemodinámica se ha llevado a cabo mediante cateterización de la arteria pulmonar. Sin embargo, el empleo de la ecocardiografía ha mejorado sustancialmente esta evaluación dirigiéndola hacia un mayor dinamismo47. Además de la evaluación de la fracción de eyección (FE) en la ecocardiografía, se podría incluir otros parámetros de cara a titular la fluidoterapia, de soporte vasoactivo o inotrópico48. Se propone que estos sean, además de la FE del ventrículo izquierdo (VI), el área del VI, el VTI aórtico (integral velocidad-tiempo del flujo transaórtico), la colapsabilidad de la vena cava inferior, el área de los ventrículos izquierdo y derecho al final de la diástole y evaluación de la función diastólica del VI mediante el estudio de las ondas E’, e’ y S. Estos, junto con parámetros clínicos como la frecuencia cardiaca, la presión arterial sistólica y diastólica y la dosis de catecolaminas, pueden diferenciar 5 fenotipos cardiovasculares macrohemodinámicos en el shock séptico: pacientes con resucitación óptima, pacientes con disfunción del VI, pacientes hipercinéticos, pacientes con disfunción del ventrículo derecho y pacientes con hipovolemia persitente49,50. Esta aproximación diagnóstica con ecocardiografía desde el inicio del soporte al paciente séptico se ha evaluado como la opción macrohemodinámica de futuro51. Una de las líneas actuales de investigación es la restricción de la resucitación con fluidos en favor del uso precoz de catecolaminas52, por lo que la evaluación mediante estos 5 fenotipos puede ayudar en la categorización de los pacientes y en estudios que conduzcan a una mejora de los desenlaces clínicos. No solo el exceso de fluidos genera resultados adversos53, sino que la administración muy precoz de vasoconstrictores no indicados puede conducir a una hipoperfusión tisular secundaria a vasoconstricción excesiva54. Por otra parte, la congestión venosa induce alteraciones en los patrones de flujo venoso en los órganos abdominales, y se puede evaluar a pie de cama mediante imagen por ecografía Doppler. Estas alteraciones se han asociado de manera consistente con la disfunción orgánica congestiva y desenlaces clínicos adversos55. En conjunto, la ecocardiografía y Doppler del sistema venoso podrían ser de utilidad para establecer un límite a la administración de fluidos en la reanimación inicial y en etapas posteriores durante el tratamiento hemodinámico de la sepsis. Sin embargo, incluso con la ecografía, la disfunción endotelial y el aumento de la permeabilidad vascular suponen un reto importante, debido a que la vida media de los fluidos administrados disminuye considerablemente y las alteraciones macrocirculatorias valoradas por ecografía podrían ser transitorias durante todo el proceso que implica la reanimación hemodinámica en sepsis.
De la miocardiopatía asociada a sepsis, se puede destacar algunas definiciones. La disfunción sistólica se ha definido en términos de FE. Sin embargo, este parámetro requiere de evaluación dinámica antes de establecer el diagnóstico. Tras un adecuado volumen de resucitación en sobrevivientes de shock séptico, se observa un aumento del gasto cardiaco y el diámetro telediastólico del VI (DTDVI) y disminución de la FE paradójicamente en comparación con los no supervivientes, en los que se observan bajos DTDVI y altas FE por resucitación inadecuada y/o sobreestimulación adrenérgica. La dilatación ventricular y la disminución de la FE corresponden a mecanismos de protección ventricular al estrés56.
La disfunción diastólica se caracteriza por un deterioro de la relajación y la distensibilidad del VI57. El estudio de la función diastólica se realiza mediante diversas técnicas de evaluación del llenado ventricular a través de Doppler transmitral, así como el grado de relajación a través del análisis del movimiento por Doppler tisular a nivel del anillo mitral lateral y medial58. La existencia de disfunción diastólica en pacientes con shock séptico se asocia con peor pronóstico59 y la aparición de hipertensión pulmonar en pacientes con neumonía y síndrome de distrés respiratorio agudo. La disfunción diastólica es favorecida por el edema miocárdico, consecuencia del aumento de la permeabilidad capilar y la extravasación proteica tisular, lo cual altera la distensibilidad cardiaca60.
La influencia de factores biológicos es fundamental en la fisiopatología de la disfunción ventricular, y se relacionan con la exposición del miocito a múltiples productos de diversa índole, producidos durante el proceso inflamatorio y daño celular (DAMP). A su vez, los procesos inflamatorios relacionados con agentes infecciosos pueden potenciar este fenómeno por medio de la producción de PAMP, como son el LPS, el ácido lipoteichoico, la flagelina y el ADN bacteriano, el manano en hongos y ARN viral de cadena simple o doble61,62.
7.2Alteraciones microcirculatorias en la sepsisLa microcirculación es la zona terminal del tejido vascular cuyo diámetro es<100μm. Las arteriolas, las venas poscapilares, capilares, vénulas, se encargan de la transferencia de oxígeno, la regulación del intercambio de solutos, el transporte hormonal y la transferencia de nutrientes. Las alteraciones en este sistema inducidas por la sepsis corresponden a una disminución en la densidad capilar y una alteración en la perfusión de la microvasculatura. Están implicadas la disfunción endotelial, la comunicación celular y la adhesividad intercelular.
La resucitación del shock séptico se ha basado convencionalmente en corregir las alteraciones de la macrocirculación a través de variables como la respuesta a la infusión de cristaloides63 o el soporte vasoactivo64. Sin embargo, existe evidencia experimental y clínica que relaciona el deterioro de la microcirculación con la disfunción orgánica y la mortalidad65. En paralelo a las alteraciones macrocirculatorias, que han sido objeto de estudio en el concepto “early goal-directed therapy”66, existen alteraciones microcirculatorias que afectan a la perfusión y están relacionadas con el desarrollo de disfunción multiorgánica y mortalidad. De la misma manera que en el shock distributivo de origen séptico, estas alteraciones se han descrito en el síndrome de respuesta inflamatoria sistémica secundario a shock cardiogénico67,68, hemorrágico69 y cirugía mayor70 (clásicamente, la cirugía cardiaca)71.
Los modelos experimentales han mostrado que las alteraciones que la sepsis ocasiona en el sistema microvascular son la disminución de la capilaridad y su densidad72 y la variabilidad en la perfusión73. Estos modelos se han estudiado en diferentes situaciones mediante la administración de endotoxina y el efecto se ha evaluado en diferentes tejidos como la piel74, la lengua, el tejido esplácnico75, el hígado o incluso tejido cerebral76.
Debido a que la alteración en la microcirculación es principalmente adaptativa, es importante investigar cómo la sepsis puede llegar a producir estos cambios. En condiciones experimentales, se ha demostrado que la hipoxia tisular, definida por presión parcial de oxígeno disminuida en presencia de reacciones con actividad redox, produce hipoperfusión en el tejido. La dirección de esta reacción es en este sentido y no al revés, en tanto que lo primario es la hipoxia, no la hipoperfusión77. En estudios realizados en la microcirculación sublingual, se ha demostrado que una mejoría de la hipoxia tisular conduce a una mejoría de la perfusión78,79. Es fácil asumir, entonces, que la mejor oxigenación está relacionada con la mejoría en la microcirculación y esta, a su vez, con la supervivencia80.
La adaptación del tejido microvascular de sujetos sanos se produce principalmente por 2 mecanismos: la acción del sistema simpático perivascular y la célula endotelial81. Además, los hematíes82 pueden llegar a funcionar como sensores intravasculares, de tal forma que la disminución de la concentración de la saturación de oxígeno induce liberación de NO y la consiguiente dilatación capilar. En la sepsis, los mecanismos implicados en la microcirculación, más allá de la disfunción endotelial, son el desequilibrio de la homeostasis entre sustancias vasoconstrictoras y vasodilatadoras, y la alteración de glucocáliz. Tyml et al.83 lo han demostrado simulando un síndrome de reperfusión en modelos experimentales. Estos estudios también se han llevado a cabo en pacientes sépticos, y la presencia de disfunción endotelial se ha relacionado con la gravedad de la disfunción orgánica84 y los desenlaces clínicos85.
En la sepsis, la interacción entre la superficie endotelial y las células circulantes también se encuentra alterada. El grosor del glucocáliz está profundamente alterado, así como sus propiedades reguladoras para la interacción celular que libera sustancias necesarias para el metabolismo86,87. La activación de la coagulación también altera la homeostasis capilar y la difusión88 del oxígeno entre los tejidos. Finalmente, la adhesión celular a este tejido dañado hace un papel importante. El fenómeno de rolling, tanto de leucocitos como de plaquetas, se encuentra aumentado en el paciente séptico, lo que potencia aún más la cascada inflamatoria. Además, el principal actor de la oxigenación tisular, el hematíe, se deforma por las alteraciones en la pared endotelial89, con lo cual disminuye la efectividad de esta célula para la secreción tanto de NO como de oxígeno. Esto prolonga el desajuste entre perfusión y oxigenación90.
8Coherencia hemodinámicaEn este sentido, Ince et al. proponen cómo optimizar el volumen de fluido para maximizar la capacidad de la microcirculación para transportar oxígeno a los tejidos. El concepto de “coherencia hemodinámica” se refiere a que el tratamiento general habitual para el shock se basa en mejorar el flujo capilar72,91. Sin embargo, a pesar de un flujo capilar normal, el transporte puede ser ineficaz por la existencia de alteraciones de la difusión de nutrientes y oxígeno. Los tipos de pérdida de coherencia hemodinámica se clasifican en 4 grupos (Fig. 3): heterogeneidad en la permeabilidad capilar dentro del mismo tejido, hemodilución que incrementa la dificultad para la difusión de sustancias, aumento de resistencias vasculares inducidas u obstrucción al flujo de retorno venoso, y edema tisular secundario al aumento de permeabilidad capilar con difusión de sustratos afectada.

Alteraciones de la microcirculación asociadas con la pérdida de la coherencia hemodinámica. Las alteraciones que conllevan la pérdida de la coherencia hemodinámica conducen a hipoxia tisular. Heterogeneidad capilar: la presencia de capilares permeables al lado de otros obturados resulta en una oxigenación heterogénea del tejido. Hemodilución: la resucitación lleva a la hemodilución, y ello hace que haya menor proporción de hematíes, por lo que hay menor proporción de transporte de oxígeno. Constricción/taponamiento: las sustancias inflamatorias alteran la regulación del tono vascular, que altera el flujo capilar y, por ende, genera anormalidades de la presión hidrostática y oncótica que conllevan una alteración en la difusión del oxígeno tisular. Edema: secundario a una situación de fuga capilar, acaba por empeorar el transporte de oxígeno por alteración de la correcta circulación del hematíe.
El objetivo del tratamiento debe ser restablecer la coherencia hemodinámica. Por lo tanto, los futuros estudios deben tener este aspecto como objetivo, centrándose en los efectos fisiopatológicos en la microcirculación. Una estrategia basada en la administración de agentes vasopresores e inotrópicos, y transfusión de hemoderivados, no tendrá el mejor resultado si no se logra mejorar y mantener el flujo en el territorio microvascular92. Este aspecto se ha demos trado en ensayos clínicos93. Dubin et al. demostraron que el aumento de la presión arterial media por encima de 65mmHg con dosis crecientes de noradrenalina puede mejorar el gasto cardiaco, la presión pulmonar, las resistencias vasculares sistémicas y los diversos parámetros macrohemodinámicos. Sin embargo, esto no es capaz de mejorar la perfusión microvascular e incluso puede ser perjudicial94, lo cual lo convierte en un factor pronóstico95. El reclutamiento de la microcirculación se relaciona con la mejoría de la función orgánica96.
En los últimos años, la atención a la respuesta microvascular se ha centrado en las intervenciones farmacológicas, con la esperanza de encontrar tratamientos capaces de restaurar el flujo microcirculatorio. La selección adecuada de los pacientes para este tipo de intervenciones es fundamental. A pie de cama, las alteraciones como la piel moteada, el tiempo de llenado capilar prolongado o el gradiente de temperatura de central a periférica son manifestaciones clínicas frecuentes de la alteración en la microcirculación97. Estos parámetros se correlacionan con la gravedad de la disfunción orgánica y predicen la mortalidad. También pueden constituir una guía de tratamiento. El relleno capilar se puede utilizar para guiar la resucitación98.
Hay autores que incluso promulgan dejar de lado los objetivos macrohemodinámicos para centrarse en la microcirculación99. Estudios experimentales muestran que las terapias vasodilatadoras pueden ser útiles en el tratamiento de las alteraciones de la microcirculación100. En pacientes con shock séptico, De Backer et al.80 demostraron como la administración tópica de acetilcolina, un agente vasodilatador endotelial, restaura la microcirculación sublingual, lo que indica un posible beneficio de las terapias vasodilatadoras.
Estos hallazgos apuntan a que el fenómeno de vasoconstricción inapropiada puede ser perjudicial y constituir una nueva diana terapéutica101. El efecto vasodilatador de la dobutamina puede resultar de utilidad como medida para reclutar la microcirculación102. También se han obtenido resultados prometedores con análogos de la prostaciclina103,104, que son potentes vasodilatadores arteriolares que pueden cumplir el criterio de actuar predominantemente en la red precapilar. Sin embargo, también hay datos contradictorios. En un ensayo aleatorizado, la administración de nitroglicerina en shock séptico no obtuvo beneficio105.
Existen diversas estrategias para seleccionar a los pacientes candidatos a estos tratamientos, así como para monitorizar la respuesta a estos106. Por una parte, la exploración clínica (moteado en piel, gradiente de temperatura) tiene la ventaja de que no requiere tecnología y es rápidamente aplicable. Sin embargo, depende del observador y se limita a la observación de la piel. La videomicroscopia, que valora tanto el flujo microvascular como la densidad capilar y la heterogeneidad de la perfusión, es actualmente el patrón de referencia para el diagnóstico. Se aplica a pie de cama, aunque requiere experiencia previa y los resultados no están disponibles inmediatamente107. La microcirculación también se puede evaluar de manera indirecta. La medición de la presión parcial de dióxido de carbono tisular (mediante capnometría sublingual o gástrica) informa sobre el estado metabólico y se puede aplicar a la cabecera del paciente, aunque es una técnica compleja. La prueba de oclusión de una extremidad permite, mediante electrodos transcutáneos, medir la presión parcial de oxígeno. Sin embargo, al ser una medida derivada de la microvasculatura, puede tener resultados dispares al evaluar diferentes territorios con
heterogeneidad de flujo. La espectroscopia infrarroja, que mide la saturación tisular de oxígeno en músculo esquelético, se puede aplicar también midiendo la respuesta a la oclusión vascular. Aunque es una medida cuantitativa, los resultados están sujetos a la heterogeneidad.
La selección de pacientes con una microcirculación profundamente alterada que no responda al tratamiento convencional es crucial108. Adecuar los tratamientos a los pacientes específicos es una estrategia obligatoria y personalizada centrada en la microcirculación. Las medidas de reanimación mayoritariamente se han orientado a la circulación sistémica. En el futuro, las investigaciones deben responder a la pregunta de si las estrategias destinadas a reclutar la microcirculación pueden mejorar los desenlaces clínicos.
8.1InmunoparálisisHistóricamente se ha hipotetizado que la fase temprana e hiperinflamatoria de la sepsis se sigue de una fase compensatoria con función antiinflamatoria que limita el daño tisular109. Los avances en el tratamiento de la sepsis han permitido acortar la duración del curso clínico, por lo que la fase compensatoria se observa con menos frecuencia. Sin embargo, existe un subgrupo de pacientes con necesidades de ventilación mecánica o fármacos vasoactivos a mediano plazo. Este estadio corresponde al proceso fisiopatológico conocido como inmunoparálisis, que se explica por la senescencia del sistema inmunitario.
La inmunoparálisis es el resultado del desequilibrio entre la respuesta innata y la adaptativa. Los intervinientes en este proceso son las células del sistema monocito-macrófago, el sistema mayor de histocompatilidad (HLA)-DR-II, las CPA en su conjunto e incluso los linfocitos T110. En los pacientes sépticos existe una regulación a la baja en la transcripción de la expresión de HLA-DR, sobre todo en los monocitos, que se ha asociado con la gravedad del curso clínico en la sepsis, la disfunción orgánica y el pronóstico. El factor clave en esto último es la vulnerabilidad a contraer infecciones secundarias o nosocomiales111.
La inmunoparálisis se puede identificar mediante el estudio de la expresión de HLA-DR en el monocito (mHLA- DR). Diversos estudios lo relacionan con una mayor mortalidad a corto y largo plazo en el shock séptico112. El HLA-DR no solo sirve como marcador, sino como guía para futuros tratamientos basados en la restauración del funcionamiento del sistema inmunológico113. La expresión reducida del HLA-DR en monocitos, que se define con un valor de HLA-DR<30%, se ha propuesto como marcador diagnóstico de inmunoparálisis en pacientes críticos114. También es un criterio diagnóstico una respuesta<200pg/ml de TNFα inducida por lipopolisacárido (LPS) durante más de 5 días115.
Se están estudiando diversos agentes adyuvantes para los pacientes que se encuentran en un estado de inmunoparálisis. Entre ellos, el factor estimulador de monocitos/macrófagos (GM-CSF), IFNγ, anti-programmed death-ligand 1 (anti PDL-1) e IL-7. En un ensayo clínico, la reducción en la expresión de mHLA-DR se ha empleado para diferenciar a los pacientes a los que se administra GM-CSF. En este estudio, la intervención ha resultado en la restauración de la función inmunológica monocítica y el acortamiento de los tiempos de ventilación mecánica y estancia hospitalaria de pacientes con shock séptico116. Respecto al elemento PD-1, se expresa en las células T activadas, las células NK y las células B117. Su ligando (PD-L1) se expresa ampliamente en células hematopoyéticas y no hematopoyéticas118. La expresión de PD-1 en las células T y las células del sistema monocito-macrófago se ha visto aumentada en pacientes con shock séptico.
La vía PD-1/PD-L1 podría desempeñar un papel importante en la inmunosupresión inducida por sepsis119. También inhibe la activación, la tolerancia y la función de las células T. Andriani et al.120 mostraron que los defectos en la función inmunitaria de los pacientes con sepsis están asociados con una expresión aumentada de PD-1 o PD-L1 y pueden restaurarse mediante el tratamiento con anticuerpos dirigidos contra PD-1 o PD-L1.
9ConclusionesLa fisiopatología de la sepsis es compleja y tremendamente heterogénea. Factores propios del microorganismo causal, del foco de infección y del huésped generan diferentes fenotipos con diferentes grados de activación de los sistemas de la inflamación, la coagulación y el endotelio. En los últimos 40 años se han producido enormes avances en el conocimiento de la fisiopatología de la sepsis, y la endotoxina se ha reconocido como la molécula principal que desencadena la respuesta inflamatoria en la sepsis causada por Gram-negativos. Sin embargo, todavía no existen tratamientos que modifiquen estos cambios fisiopatológicos y se traduzcan en una mejora de la morbimortalidad de los pacientes sépticos. Las alteraciones hemodinámicas en la macrocirculación son esenciales durante el tratamiento de la sepsis a pie de cama. Sin embargo, en los últimos años se ha puesto el foco en las alteraciones de la microcirculación y la resucitación con fluidos utilizando medios prácticos, como por ejemplo la ecografía, siempre tomando en cuenta objetivos basados en restaurar la pérdida de la “coherencia hemodinámica”. La heterogeneidad de la sepsis hace necesaria una mejor selección de pacientes en futuros ensayos clínicos, con base en los diferentes fenotipos, lo que probablemente facilitará el hallazgo de beneficios de estos esperados tratamientos.
10FinanciaciónNo fue necesaria financiación.
11Conflicto?de?interesesLos autores no declaran conflictos de intereses en la realización de este trabajo.
12Nota al suplementoEste artículo forma parte del suplemento «Resucitación del paciente con sepsis y shock séptico», que cuenta con el patrocinio de AOP HEALTH IBERIA.

