


INTRODUCCIÓN
Los términos "muerte celular programada" y "apoptosis" suelen utilizarse indistintamente para definir un tipo de muerte celular, diferente de la necrosis, que regula de forma fisiológica la homeostasis de un organismo. La apoptosis celular es la base de muchos procesos fisiológicos y su conocimiento ha progresado rápidamente en la última década. Los que inicialmente fueron únicamente hallazgos de laboratorio están adquiriendo, cada vez más, una relevancia clínica. Esta revisión pretende el acercamiento del médico intensivista a los conceptos básicos y a los hallazgos más relevantes descritos hasta ahora sobre la apoptosis en el campo de la Medicina Intensiva. Para facilitar su lectura se ha considerado la apoptosis por órganos y tipos de patologías, asumiendo que esta compartimentalización puede ser artificiosa en muchas ocasiones.
La necrosis celular siempre se ha considerado perjudicial para el organismo. En la muerte por necrosis la célula se hincha, pierde la integridad de su membrana y libera el contenido celular. Este hecho da lugar a una respuesta inflamatoria, iniciada fundamentalmente por las enzimas proteolíticas y los radicales libres que la célula muerta albergaba en su interior1. Por el contrario, la célula apoptótica presenta una balonización de su membrana plasmática, la cromatina nuclear se condensa y disminuye su volumen. Además, diferentes mecanismos activan endonucleasas que degradan el ADN a nivel de los nucleosomas originando el típico patrón en escalera al migrar en un gel de electroforesis2, 3. Así, la célula apoptótica es rápidamente fagocitada y digerida por células fagocíticas vecinas, con lo cual no se libera su contenido y no se originan fenómenos inflamatorios4. Kerr et al, al observar estos cambios morfológicos en estudios con embriones, fueron los primeros en utilizar el término apoptosis (del griego appo-toe-sis, indicativo de la caída de las hojas de un árbol o de los pétalos de una flor)2. A los hallazgos morfológicos le siguieron los estudios moleculares en el gusano Caenorhabditis elegans, donde por primera vez se evidenció la existencia de un control genético en la apoptosis celular. Estos trabajos fueron motivo de la concesión del Premio Nóbel de Medicina en 2002. Actualmente los cambios morfológicos de la apoptosis celular se estudian mediante microscopía electrónica y es posible cuantificar la apoptosis por técnicas enzimáticas, inmunohistoquímicas, citométricas y moleculares.
MECANISMOS MOLECULARES DE LA APOPTOSIS
La muerte celular por apoptosis es un proceso regulado genéticamente, en el cual intervienen moléculas efectoras y moléculas reguladoras de la apoptosis. Muchas de las señales de muerte y supervivencia celular proceden del exterior de la célula a través de receptores de superficie (vías de muerte extrínsecas). Otro mecanismo de inducción de apoptosis es el intrínseco, ante la falta de factores de crecimiento o inducidos por alteración intrínseca de mecanismos reguladores de apoptosis5.
Vía de muerte celular extrínseca
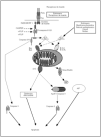
La muerte celular por apoptosis puede iniciarse mediante la unión de un ligando de muerte a un receptor especializado (receptores de muerte) en la membrana plasmática de la célula. Los receptores de muerte pertenecen a la superfamilia de genes del receptor del factor de necrosis tumoral (TNFR). De ellos, el mejor conocido en cuanto a inducción de apoptosis es Fas, el cual se activa al unirse a su ligando (FasL). Estos receptores son proteínas, que se expresan en muchas células del organismo, y presentan a nivel intracelular una secuencia citoplasmática denominada dominio de muerte6, 7. Cuando el receptor de membrana se activa al unirse a su ligando se produce una serie de alteraciones en el dominio de muerte que conlleva un reclutamiento de moléculas adaptadoras que permiten la activación de las caspasas iniciadoras. Esas moléculas adaptadoras son TRADD (dominio de muerte asociado al TNFR) y FADD (dominio de muerte asociado a Fas). Los dominios de muerte asociados a Fas o a TNFR normalmente están inactivos en forma de monómeros. Cuando los ligandos se unen a esos receptores, los dominios de muerte son trimerizados y ya pueden activar a las caspasas iniciadoras y ejecutar el programa de muerte8-10 (fig. 1).

Figura 1. Vías de muerte celular programada. Se representan las principales vías de apoptosis. FasL: ligando de Fas; FADD: dominio de muerte asociado a Fas; cit C: citocromo C; IAP: proteína inhibidora de apoptosis; AIF: factor inductor de apoptosis; Smac/Diablo: Second mitochondria-derived activator of caspases/ Direct IAP-binding protein with low pI.
Las caspasas son una familia de cisteín proteasas fielmente conservadas a lo largo de la evolución. Estas proteasas se denominan así por la presencia de un residuo de cisteína en su centro catalítico y por hidrolizar las proteínas detrás del aminoácido aspartato. Las caspasas se sintetizan como precursores inactivos compuestos por diferentes dominios o regiones peptídicas: un dominio amino terminal de tamaño variable, una subunidad larga, una subunidad corta y una región de unión entre ambas subunidades. La activación de las caspasas supone la fase efectora de la apoptosis y se puede producir por numerosos estímulos. Uno de ellos es la activación de la caspasa 8 por la unión de factores solubles (FasL y TNF) a sus receptores de membrana (fig.1). Otro mecanismo regulador de las caspasas es la familia de Bcl-211.
La activación de cada caspasa se induce por digestión proteolítica entre dominios y por el posterior ensamblaje de las subunidades larga y corta para formar la enzima activa. Las caspasas se clasifican en iniciadoras (caspasas 1, 2, 4, 5, 8, 9 y 10) que tienen prodominios largos, de 15-25 kd, y en efectoras (caspasas 3, 6 y 7) que tienen prodominios de menos de 5 kd.
El desmantelamiento estructural característico de una célula por apoptosis es un proceso complejo, que implica el procesamiento de un gran número de sustratos celulares por las caspasas efectoras. Uno de los sustratos fundamentales de estas proteasas es la nucleasa responsable de la degradación del ADN. Esta nucleasa corta el ADN genómico entre nucleosomas generando fragmentos de tamaño múltiplo de 180 pares de bases, originándose un patrón en escalera en un gel de agarosa característico de la apoptosis. Esta nucleasa de ADN se denomina Cad (Caspase-Activated DNase) y su activación se produce a través de la proteólisis de Icad mediada por la caspasa 3, que permite la liberación y activación de la subunidad catalítica (fig. 1).
Vía de muerte celular intrínseca. Familia de Bcl-2
El otro mecanismo principal de inducción del programa de apoptosis es el intrínseco, a partir de señales intracelulares (fig. 1). Esta vía participa fundamentalmente en aquella muerte celular inducida por deprivación de factores de crecimiento, radiaciones, corticoides, etc. De la vía intrínseca depende la apoptosis de células durante el desarrollo embrionario y la homeostasis celular en condiciones fisiológicas. Existe una familia de proteínas esencial en la regulación de la vía intrínseca de apoptosis, conocida como familia de Bcl-2, por ser éste el primer miembro descrito. El gen Bcl-2 fue identificado por primera vez en un linfoma de células B-2, motivo de su denominación. La familia se compone al menos de 16 miembros12 (fig. 2). Consta de una variedad de genes agrupados en función de sus características estructurales y funcionales. Entre ellos existen algunos que inhiben la apoptosis y otros que la inducen. Los diferentes miembros de la familia de Bcl-2 se encuentran principalmente en la mitocondria o en el citosol de la célula y forman dímeros entre sí. Los niveles relativos de miembros inductores e inhibidores de la apoptosis funcionan como un reostato regulando el umbral apoptótico de la célula. Así, por ejemplo, las proteínas antiapoptóticas Bcl-2 y Bcl-XL forman heterodímeros con la proteína proapoptótica Bax, de tal suerte que un exceso de Bax facilitará la muerte celular por apoptosis al incrementar los homodímeros Bax/Bax proapoptóticos, y un exceso de Bcl-2 o Bcl-XL darán lugar al efecto contrario al generar homodímeros Bcl-2/Bcl-2 o Bcl-XL/Bcl-XL y la célula sobrevivirá.
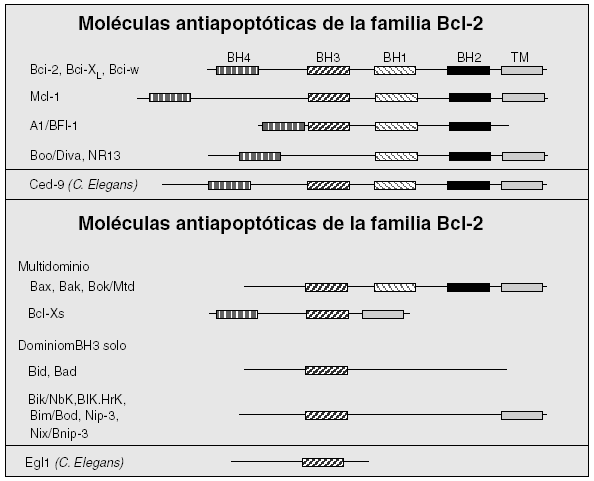
Figura 2. Miembros de la familia de Bcl-2. Los miembros de la familia de Bcl-2 poseen hasta 4 dominios de estructura en hélice de homología (denominados desde BH1 hasta BH4) que se representan por rectángulos. Parte de los miembros se anclan a membranas a través de un dominio transmembranal (TM). Se observa cómo los miembros antiapoptóticos se caracterizan por conservar los 4 dominios BH. Por el contrario, los proapoptóticos se dividen en los que conservan la mayoría de los dominios BH (multidominio) y los que sólo conservan el dominio BH3, junto con la región TM o no. Además, se muestran los homólogos en el nematodo Caenorhabditis elegans (C. elegans) de los miembros de la familia de Bcl-2.
Estudios de mutagénesis han demostrado que los dominios BH1, BH2 y BH3 están implicados en los procesos de homo y heterodimerización. La capacidad de inducir apoptosis por parte de los miembros proapoptóticos puede realizarse mediante la inhibición de miembros antiapoptóticos, a través del dominio BH3, o mediante la capacidad de estas proteínas de insertarse en la membrana mitocondrial y formar poros en dicha membrana. De esta forma, la permeabilización de la membrana exterior de la mitocondria, como parte de la disfunción mitocondrial, se acompaña de la liberación del citocromo C, Smac/Diablo y del factor inductor de la apoptosis (AIF). El citocromo C es un componente de la cadena transportadora de electrones necesaria para la respiración mitocondrial. Cuando se libera dentro del citoplasma, el citocromo C induce la oligomerización de Apaf1, y como resultado se activa la caspasa 9 y seguidamente la caspasa 3 (fig. 1).
Existen otras vías de apoptosis como la vía de la perforina-granzima B, utilizada por los linfocitos citotóxicos; la vía de la esfingomielasa, etc. No se va a profundizar en ellas por escaparse de los objetivos de esta revisión.
PRINCIPALES DISFUNCIONES ORGÁNICAS Y APOPTOSIS
Son innumerables las patologías en las que se ha demostrado una alteración de la apoptosis, bien por exceso bien por defecto4. No obstante, en muchas ocasiones no puede asegurarse que el hallazgo de apoptosis en un proceso patológico sea importante en la patogenia o en la evolución del mismo. A continuación se comentan agrupadas por sistemas algunas de las patologías críticas en las que se han hallado resultados de interés.
Sistema inmunitario
Hasta épocas muy recientes se concebía la sepsis como una respuesta inflamatoria sistémica e incontrolada del organismo a la infección13. Sin embargo, a partir del fracaso de la gran mayoría de los ensayos clínicos realizados con medicamentos que bloqueaban la cascada inflamatoria, se ha observado que la respuesta a la infección no es lineal y que existe un perfil inflamatorio variable de unos individuos a otros y también dentro del mismo individuo14. Ante el estímulo de un patógeno, el organismo reacciona activando el sistema inmune, incluyendo a los macrófagos, células dendríticas y linfocitos T CD4. Los macrófagos y las células dendríticas se activan por la ingestión del patógeno y por la estimulación de las citocinas proinflamatorias secretadas por los CD4. Sin embargo, estos CD4 también pueden liberar citocinas antiinflamatorias que suprimen la activación de los macrófagos, y pueden ser, así mismo, activados por los macrófagos y las células dendríticas. Dependiendo de diversos factores como el patógeno causante de la infección, el sitio de infección, etc., los macrófagos y las células dendríticas responderán favoreciendo la producción de citocinas pro o antiinflamatorias, o favoreciendo una reducción global de la función del sistema inmunitario14. La apoptosis celular puede favorecer esta depresión inmunitaria producida por la sepsis15. Diversos estudios experimentales han demostrado que durante la sepsis los linfocitos y las células del epitelio gastrointestinal mueren por apoptosis16, 17. Estos hallazgos fueron refrendados en un estudio autópsico que demostró que los pacientes fallecidos por sepsis tenían una tasa de apoptosis muy superior a los que lo hicieron por otra causa en las células linfocíticas y del epitelio gastrointestinal18. Los linfocitos y el epitelio gastrointestinal tienen una elevada tasa de recambio celular mediada por apoptosis y, probablemente la sepsis, acelera ese proceso natural15.
Actualmente existe evidencia de que la inhibición de la apoptosis podría estar desempeñando un importante papel en la reducción de la mortalidad que algunos tratamientos han demostrado recientemente en pacientes con sepsis grave. El efecto beneficioso de la proteína C activada en la sepsis grave19 podría no deberse únicamente a su efecto antitrombótico, ya que otros medicamentos anticoagulantes fracasaron previamente. Joyce et al20 han demostrado recientemente que la proteína C previene la apoptosis en diversas líneas celulares humanas, incluyendo las células endoteliales, favoreciendo la expresión de genes antiapoptóticos y disminuyendo la expresión de genes proapoptóticos. Además, estimula la actividad de la óxido nítrico sintetasa endotelial21.
El estudio de los mecanismos intrínsecos antiinflamatorios de los corticoides ha revelado que estos fármacos retrasan la apoptosis de los granulocitos e incrementan la fagocitosis de las células apoptóticas por los macrófagos, procesos que pueden ser esenciales para el control y resolución de la inflamación22, 23. Finalmente, también se han descrito efectos antiapoptóticos para la insulina24, que podrían explicar en parte, su efecto protector en el paciente séptico25.
La inhibición de la apoptosis celular durante la sepsis es una posibilidad terapéutica interesante. En modelos experimentales de sepsis Hotchkiss et al demostraron que la prevención de la apoptosis linfocitaria mejoraba la supervivencia26. También evidenciaron que la sobreexpresión de Bcl-2 en células T de ratones transgénicos disminuía la apoptosis linfocitaria e incrementaba la supervivencia27. Otros estudios experimentales han sugerido que la inhibición de apoptosis intestinal que se produce en la sepsis, mediante la sobreexpresión de Bcl-2 mejora la supervivencia en diferentes modelos de sepsis28, 29.
Los estudios con pacientes son muy escasos hasta la fecha, intentando sobre todo confirmar aspectos fisiopatológicos observados en estudios experimentales. Martins et al observaron en pacientes sépticos un incremento de la capacidad fagocítica de los neutrófilos y una posterior pérdida de neutrófilos por apoptosis que justificaron como un mecanismo regulador de la respuesta inflamatoria30. Aún así, estos resultados deben ser evaluados con cautela porque existen trabajos contradictorios31. Los mecanismos responsables de estos hallazgos no son plenamente conocidos. El tratamiento de los pacientes sépticos con inhibidores de la apoptosis es muy controvertido y plantea diversos problemas, entre ellos el control de la inhibición sobre un único grupo celular. Recientemente, Cauwels et al demostraron que el bloqueo indiscriminado de caspasas puede empeorar el pronóstico en modelos experimentales de sepsis. La inhibición general de caspasas incrementó la toxicidad in vivo inducida por el TNF produciendo un incremento en el estrés oxidativo, dañando la mitocondria e incrementando la mortalidad32. Actualmente existen diversos estudios en fase experimental y preclínica con inhibidores de las caspasas, en particular la caspasa 333.
El tratamiento de la sepsis mediante bloqueo o inducción de la apoptosis es un aspecto novedoso de la terapéutica de la principal causa de mortalidad en las Unidades de Cuidados Intensivos (UCI). Es necesario profundizar en el conocimiento de la respuesta inflamatoria y del papel que tiene la apoptosis.
Sistema nervioso central
El estudio de los mecanismos de apoptosis durante la embriogénesis y la homeostasis celular del sistema nervioso central (SNC) en la vida adulta ha sido facilitado, en gran medida, por el conocimiento de la muerte celular programada en otros sistemas celulares y tisulares34. Pero este tipo de muerte celular no participa sólo en la fisiología del SNC. En los últimos años han aparecido trabajos que muestran cómo la muerte neuronal apoptótica contribuye al desarrollo de alteraciones neurodegenerativas del SNC, como el Parkinson35, 36, enfermedad de Hungtinton, epilepsia, enfermedad de Alzheimer35, 37 y en trabajos experimentales de isquemia cerebral38, 39. A raíz de estos trabajos, en la última década se comenzó a estudiar la posible implicación de la muerte por apoptosis en el daño cerebral agudo (DCA), tanto por traumatismo craneoencefálico (TCE), como por hemorragia cerebral (HC) o ictus isquémico. Rink et al, en 1995, describieron por primera vez la presencia de neuronas apoptóticas en modelos experimentales de cerebros de ratas sometidos a un TCE40. Estos hallazgos se han observado recientemente en cerebros humanos sometidos a un DCA41,42.
Los trabajos mencionados han impulsado los estudios encaminados a determinar la importancia que puede tener la apoptosis neuronal en el pronóstico de los pacientes con un TCE. Así, ha aumentado la evidencia acerca del papel decisivo que tiene la familia de Bcl-2 en la regulación de la apoptosis en pacientes que sufren dicho tipo de traumatismo. Raghupathi et al demostraron que la sobreexpresión de Bcl-2 humano en ratones transgénicos disminuía el daño a nivel neuronal y se reducían las secuelas motoras tras inducirse un TCE43. Estos resultados fueron refrendados por nuevos estudios experimentales que confirmaron el papel neuroprotector de la sobreexpresión de la proteína Bcl-2 en modelos experimentales de TCE44. En un estudio con 11 pacientes a los que se practicó una craniectomía para extirpar las áreas de contusión cerebral debidas a un TCE, se demostró que la expresión del gen Bcl-2 en las áreas pericontusionales se asociaba con una mayor supervivencia de los pacientes45. Otros trabajos han demostrado, tanto en cerebros de ratas46-49 como de seres humanos42 sometidos a un TCE, que existe una regulación alterada de la familia Bcl-2 y de las caspasas tras una lesión cerebral.
El cerebro humano es capaz de sintetizar factores solubles tras un TCE50-52. De hecho, se ha observado cómo la acumulación de componentes séricos extravasados en la zona de edema cerebral se acompaña de muerte celular por apoptosis en las células circundantes53. Por ello, nuestro grupo se ha centrado en el estudio de factores solubles obtenidos a partir del suero drenado del bulbo de la vena yugular interna en pacientes que sufrieron un TCE, analizando, de esta forma, los posibles factores solubles sintetizados a nivel cerebral tras un TCE grave. Así, hemos demostrado que el suero obtenido del bulbo de la vena yugular interna (regional) de pacientes que han sufrido un TCE es capaz de inducir apoptosis in vitro de células linfoides54, 55. La apoptosis observada podría estar inducida por la expresión aumentada de una proteína proapoptótica (Bax) y la disminución de la expresión de una proteína antiapoptótica (Bcl-XL). Asimismo, la tasa de apoptosis observada se ha correlacionado con la mortalidad y la situación funcional de los pacientes a los 6 meses del TCE54 (manuscrito enviado para publicación).
Se han realizado experimentos con diversos medicamentos inhibidores de la apoptosis en modelos experimentales de TCE. Siren et al demostraron que la administración de eritropoyetina en ratas sometidas a una oclusión de la arteria cerebral media disminuyó el número de neuronas que presentaron apoptosis y, paralelamente, la extensión del infarto cerebral56. Este mismo grupo de trabajo demostró que el pretratamiento con eritropoyetina disminuía la apoptosis en cultivos neuronales a los que se retiraron los nutrientes56. Nuevos estudios han refrendado estos resultados57. Otros autores han mostrado que el tratamiento con inhibidores de la caspasa 3 mejora el pronóstico en modelos experimentales de TCE58 y de infarto isquémico cerebral59.
La lesión cerebral traumática es uno de los campos de la Medicina Intensiva donde mayores avances se han producido en relación con el papel de la muerte celular programada. Sin embargo, a pesar de los buenos resultados obtenidos a nivel experimental, son necesarios más estudios que permitan confirmar en seres humanos la posibilidad de utilizar, en un futuro, bloqueadores de la apoptosis como terapia en pacientes con un TCE.
Sistema respiratorio
La lesión pulmonar aguda (LPA) se asocia a una rotura del epitelio alveolar, de la membrana basal y del endotelio, lo que produce un incremento de la permeabilidad del epitelio pulmonar60. Existe un gran interés en conocer el papel de los mediadores inflamatorios en la patogénesis del daño epitelial y endotelial de la LPA. En los últimos tiempos se ha comenzado a estudiar el papel que desempeña la apoptosis en los pacientes con LPA61.
La LPA y el síndrome de distrés respiratorio agudo (SDRA) pueden agravarse por los neutrófilos que liberan proteasas y radicales libres. La neutrofilia característica del SDRA puede deberse a una alteración en la migración, a un exceso de activación o a un descenso en la apoptosis de los neutrófilos. Los mediadores inflamatorios como citocinas, la hipoxia y la acidosis activan a los neutrófilos y retrasan su apoptosis. No se conocen completamente los mecanismos que producen la inflamación alveolar en la LPA o en el SDRA, pero la apoptosis de los neutrófilos podría ser un importante mecanismo regulador, permitiendo la eliminación de los neutrófilos del área afectada con un mínimo daño pulmonar62, 63. Los neutrófilos se eliminan del pulmón por macrófagos y otras células fagocíticas. El receptor del macrófago CD44 parece tener un papel fundamental en el aclaramiento de los neutrófilos apoptóticos in vivo e in vitro64, 65. En modelos experimentales de LPA con ratones transgénicos carentes de CD44, se ha observado que el aclaramiento de neutrófilos disminuía de forma muy importante, aumentando la inflamación pulmonar y la tasa de mortalidad de los ratones65. Sin embargo, existen muchas lagunas en el conocimiento de estos procesos. La apoptosis de los neutrófilos puede retrasarse por factores como los factores estimulantes de colonias monocito-macrófago (GM-FSC). En un estudio realizado por Matute-Bello et al se observó que el líquido broncoalveolar (LBA) de pacientes con SDRA tenía concentraciones elevadas de GM-FSC. Se vio que los pacientes con SDRA que sobrevivieron tenían una mayor tasa de GM-FSC que los fallecidos66. Estos autores concluyeron que en ciertos casos (como el SDRA asociado a infecciones bacterianas) la inhibición de la apoptosis de los neutrófilos podía ser beneficiosa. Estos hallazgos aumentan la discrepancia entre los autores acerca del efecto beneficioso o nocivo de la apoptosis en la LPA y en el SDRA.
Otra de las características del SDRA es la destrucción del epitelio alveolar y la afectación del endotelio en los capilares alveolares. Diversos estudios han demostrado que en pacientes con SDRA el mantenimiento de la función epitelial se asocia con un mejor pronóstico67, 68. Bachofen et al mostraron en pacientes fallecidos en el curso inicial del SDRA que los neumocitos tipo I morían por apoptosis69. Estudios posteriores evidenciaron un incremento de Bax (gen proapoptótico) en las células del epitelio alveolar70. Otros autores han demostrado que la expresión de Fas está incrementada en pacientes fallecidos por SDRA. Asimismo, FasL puede cuantificarse en el líquido del edema pulmonar de pacientes con LPA71.
Se desconocen muchos aspectos de la apoptosis en la LPA y el SDRA; de hecho, muchos estudios son contradictorios en cuanto al beneficio o perjuicio en este tipo de pacientes. Existen muchas evidencias, sobre todo experimentales, sobre el importante papel que tienen los neutrófilos en la patogenia de la LPA y el SDRA. Es posible que el hipotético beneficio de la manipulación de la apoptosis en la patología pulmonar grave dependa del tipo de células afectadas y del momento de la evolución del daño pulmonar. Son necesarios más estudios que permitan conocer los aspectos fisiopatológicos de la apoptosis en la LPA y en el SDRA para, en un futuro, plantearse aplicar terapias específicas a estas patologías.
Sistema cardiovascular
Se ha demostrado, tanto en estudios in vitro como en modelos animales in vivo, que se produce apoptosis en los miocitos tras un infarto agudo de miocardio (IAM), especialmente en la periferia de un IAM donde la isquemia ha sido menor72. El manejo precoz del IAM con trombolíticos o angioplastia restaura el flujo sanguíneo y minimiza la necrosis miocárdica. Sin embargo, en la reperfusión miocárdica se libera una gran cantidad de radicales libres que producen muerte celular por apoptosis. De hecho, diversos trabajos experimentales han demostrado que el uso de bloqueadores de la apoptosis mejora el pronóstico en el IAM. Así, Parsa et al mostraron que el uso de eritropoyetina en cultivos in vitro de cardiomiocitos y administrado en conejos in vivo disminuía la apoptosis de los cardiomiocitos y con ello la extensión del IAM, con lo que se mejoraba la función ventricular73. Nuevos estudios experimentales han confirmado dichos resultados74. Es posible que en un futuro el mejor conocimiento de los mecanismos apoptóticos permita disminuir el daño producido tras la reperfusión miocárdica.
En los vasos sanguíneos, además de la importancia que la apoptosis pueda tener en el remodelamiento vascular, se han realizado estudios en la aterosclerosis. Las células inflamatorias y los miofibroblastos que se observan en la placa de ateroma pueden eliminarse mediante apoptosis. Es posible que una apoptosis mal regulada en la zona de ateroma contribuya a la progresión de la enfermedad75.
Sistema gastrointestinal
El intestino es, en teoría, susceptible a cambios apoptóticos dada su elevada tasa de recambio celular. De forma tradicional se ha considerado que la integridad del epitelio intestinal tiene un papel muy importante en la sepsis evitando la translocación bacteriana76. Estudios experimentales con ratas han demostrado la existencia de apoptosis en el epitelio intestinal. Mas aún, en modelos de sepsis la tasa de apoptosis se incrementa observándose un pico de apoptosis máximo en las primeras 24 horas tras el inicio de la infección. El aumento en la apoptosis se produce mediante la activación de la vía mitocondrial, dado que se ha observado que en ratones transgénicos la sobreexpresión de Bcl-2 disminuye la apoptosis intestinal77 y mejora la supervivencia28. Asimismo, en estudios con ratas sometidas a neumonía por Pseudomonas aeruginosa la sobreexpresión de Bcl-2 a nivel intestinal incrementa la supervivencia asociándose a un descenso de la apoptosis intestinal29. Hotchkiss et al demostraron en una serie de pacientes fallecidos en una UCI que la tasa de apoptosis intestinal era muy superior en los pacientes fallecidos por sepsis respecto a los fallecidos por otras causas18. No obstante, los estudios realizados, sobre todo en seres humanos, son todavía muy limitados.
Sistema renal
Aunque la necrosis es el principal mecanismo de muerte celular en el fracaso renal agudo (FRA), la muerte por apoptosis también desempeña un papel importante78. Estudios experimentales han demostrado la existencia de apoptosis en el túbulo renal, tanto en la fase de hipoxia como en la de reperfusión renal79. Además, se ha evidenciado apoptosis en los túbulos renales en biopsias de pacientes fallecidos con FRA80. El número de células apoptóticas debido a la fase de reperfusión renal se correlacionaba con la duración de la isquemia renal81. En injertos renales, obtenidos de cadáver, se ha observado que tras la lesión por isquemia y reperfusión se produce apoptosis en los túbulos renales mediante la activación de la vía mitocondrial (se observó un incremento en la expresión de Bax en el 100% de las muestras estudiadas). La tasa de apoptosis se correlacionó con el tiempo de isquemia fría82. Otros autores han demostrado que la tasa de apoptosis en las células del túbulo renal de los injertos renales de cadáver son un factor predictivo para la función precoz del riñón postransplante83. Asimismo, la disminución en la expresión de Bcl-2 en la biopsia del túbulo proximal del donante se asocia con peor función del injerto en el paciente trasplantado84. En enfermos críticos se ha demostrado apoptosis del túbulo renal asociada a sepsis mediante la activación del receptor de membrana del TNF y la posterior activación de la ruta de las caspasas85. En modelos experimentales se ha demostrado que fármacos nefrotóxicos como la gentamicina o quimioterápicos pueden producir apoptosis tanto en el túbulo proximal como en el distal86.
POSIBILIDADES TERAPÉUTICAS BASADAS EN LA REGULACIÓN DE LA APOPTOSIS
En la base de la patogénesis de cualquier enfermedad humana es frecuente que aparezca un componente apoptótico que contribuya a la progresión de la enfermedad. Por tanto, la modulación de la apoptosis como tratamiento tiene un gran potencial terapéutico. Modular la expresión de los componentes moleculares claves de la maquinaria apoptótica es una estrategia obvia mediante terapia génica. De hecho, se está estudiando la inhibición de la expresión génica con oligonucleótidos antisentido y el uso de moléculas biológicas recombinantes, sobre todo en el área de la Oncología. Sin embargo, tienen un gran número de limitaciones prácticas, especialmente en el campo de la Medicina Intensiva. El papel de la apoptosis está aún pendiente de establecerse con claridad y los procedimientos terapéuticos están en sus inicios.
En patología crítica se están utilizando diferentes fármacos que suelen tener a las caspasas como dianas terapéuticas. Se han realizado numerosos estudios experimentales con inhibidores de caspasas. En distintos modelos de isquemia (hepática, cardíaca, renal, intestinal y cerebral) la inhibición de caspasas ha resultado positiva, ya que además de disminuir la apoptosis ha mejorado la supervivencia y la función del órgano. Sin embargo, existen resultados contradictorios que demuestran que el bloqueo indiscriminado de caspasas puede empeorar el pronóstico32. Son muy prometedores los estudios en daño cerebral traumático, hemorrágico o isquémico, donde se ha evidenciado el papel decisivo que tiene la familia de Bcl-2 en el pronóstico del DCA, tanto en animales como en seres humanos. Sin embargo, todavía no se han demostrado en estos últimos los excelentes resultados obtenidos mediante la inhibición neuronal de apoptosis en diferentes modelos experimentales.
La modulación de la apoptosis ofrece unas enormes posibilidades terapéuticas en el campo de la Medicina Intensiva. Aun así, actualmente existen muchas limitaciones prácticas que dificultan su desarrollo. El conocimiento del papel que desempeña la apoptosis en la patología crítica es incompleto. Se comienzan a conocer las consecuencias que se producen al incrementar o disminuir la apoptosis en diferentes órganos; sin embargo, no está aclarado si dicho incremento o disminución son beneficiosos o perjudiciales. La modulación de la apoptosis debe ser adecuada en cada momento según las necesidades del órgano afectado. Además, para poder conseguir un efecto terapéutico beneficioso, la modulación apoptótica debe ser organoespecífica. Una activación indiscriminada de la apoptosis puede resultar perjudicial para varios órganos e incluso inducir la aparición de tumores. Por otra parte, la supervivencia de una célula por la inhibición de apoptosis no significa el mantenimiento de la función celular. Es posible que la inhibición de la apoptosis celular produzca una célula no funcional.
Sin duda alguna, nos encontramos en los inicios de una nueva posibilidad terapéutica en el campo de la Medicina Intensiva. En los años sucesivos se desarrollarán nuevos estudios que permitirán dilucidar el papel que puede tener la apoptosis en el paciente crítico.
AGRADECIMIENTOS
Al Dr Álvaro Castellanos (Servicio de Medicina Intensiva. Hospital Universitario Marqués de Valdecilla) por su ayuda en la revisión de este manuscrito.
Este trabajo ha sido financiado, en parte, por una beca FIS (n.o PI020417) y por una beca de la Fundación Marqués de Valdecilla.

