




Editado por: F. Baigorri-González y J.º. Lorente Balanza
Última actualización: Abril 2005
Más datosBásicamente la sepsis se pone en marcha cuando unos activadores procedentes de los microorganismos patógenos o de sus productos desencadenan estímulos celulares y humorales que, bien directamente o bien a través de citocinas y otros mediadores producen unos efectos biológicos que se traducen en efectos clínicos.
Estos activadores son globalmente llamados en la actualidad comportamientos moleculares asociados a patógeno o PAMP (pathogen-associated molecular patterns) y los mecanismos que ponen en marcha pueden diferir dependiendo del germen causal.
SEPSIS POR GRAMNEGATIVOS
La sepsis iniciada por gramnegativos se desencadena por el lipopolisacárido conocido como endotoxina (LPS). La LPS es vertida a la circulación donde se enfrenta a una primera línea de sustancias naturales que intentan bloquear la infección: anticuerpos, albúmina, lipoproteínas de alta intensidad (HDL) y BPI (bactericidal permeability increasing protein) expresada por polimorfonucleares (PMN), monocitos/macrófagos (M/M) y eosinófilos y, sobre todo, a través de los receptores de la respuesta del sistema inmune innato expresados por dichas células. Funcionalmente estas proteínas pueden ser divididas en tres clases: segregadas, como las opsoninas, endocíticas y de señal. La mejor estudiada es la lectina unida a manano que, al unirse a los carbohidratos microbianos, inicia la vía de la lectina para la activación del complemento.
La LPS que continúa circulante se une a la LBP (lipopolysaccharide binding protein) que es una proteína producida por el hígado. Este complejo va a unirse a los receptores de la pared celular CD14 (fundamentalmente de los macrófagos) iniciándose la secuencia de la señal intracelular a través del complejo TLR4, del que posteriormente hablaremos, y la proteína MD-2. En las células donde no existen receptores CD14 (como en las células endoteliales, células dendríticas, fibroblastos, células del músculo liso), esta cascada se inicia uniéndose el complejo LPS-LBP a CD14 soluble circulante en el plasma. Existen otros receptores de la pared celular que reconocen a la LPS como el MSR (macrophage scavenger receptor), canales de K+, y los receptores CD11/CD18. El CD14 está unido a la membrana por un anclaje glicosil-fosfatidil-inositol que carece de dominio transmembrana, ello se obvia por proteínas identificadas como receptores tipo portazgo toll-like receptors (TLR) que inician la vía de señales que implica al factor nuclear kappa-B (NF-κB) y a la subsiguiente transcripción genética.
La señal intracelular se inicia con la unión del dominio intracelular TLR llamado TIR (Toll/IL-1 receptor homology domain) a una kinasa asociada, IRAK (IL-1 receptor-associated kinase). Este proceso requiere dos proteínas de adaptación, las llamadas MyD88 (myeloid differentiation protein 88) y TIRAP (TIR domain containing adapter protein), llamada también Mal (MyD88-adapter-like protein). Y a su vez puede inhibirse por una tercera proteína llamada Tollip (Toll-intercating protein). Se produce un proceso de fosforilización y se asocia a otra proteína, TRAF6 (Tumor necrosis factor receptor-associated factor-6) que activa a otra kinasa, la MAP3K (mitogen-activated protein kinase kinase kinase) para actuar sobre el complejo IKK (inhibitor KB kinase) que precisa la proteólisis a través del sistema ubiquitina del inhibidor IκB para que se liberen los dímeros del NF-κB (RelA [p65], c-Rel, RelB, p50, y p52) en el núcleo donde se hacen activos, traslocan al núcleo y permiten la traslación, transcripción y producción de un ARNm mensajero que induzca la producción de citocinas y otras moléculas efectoras1. Teóricamente una sepsis debe persistir mientras continúe la translocación nuclear de NF-κB (fig. 1).
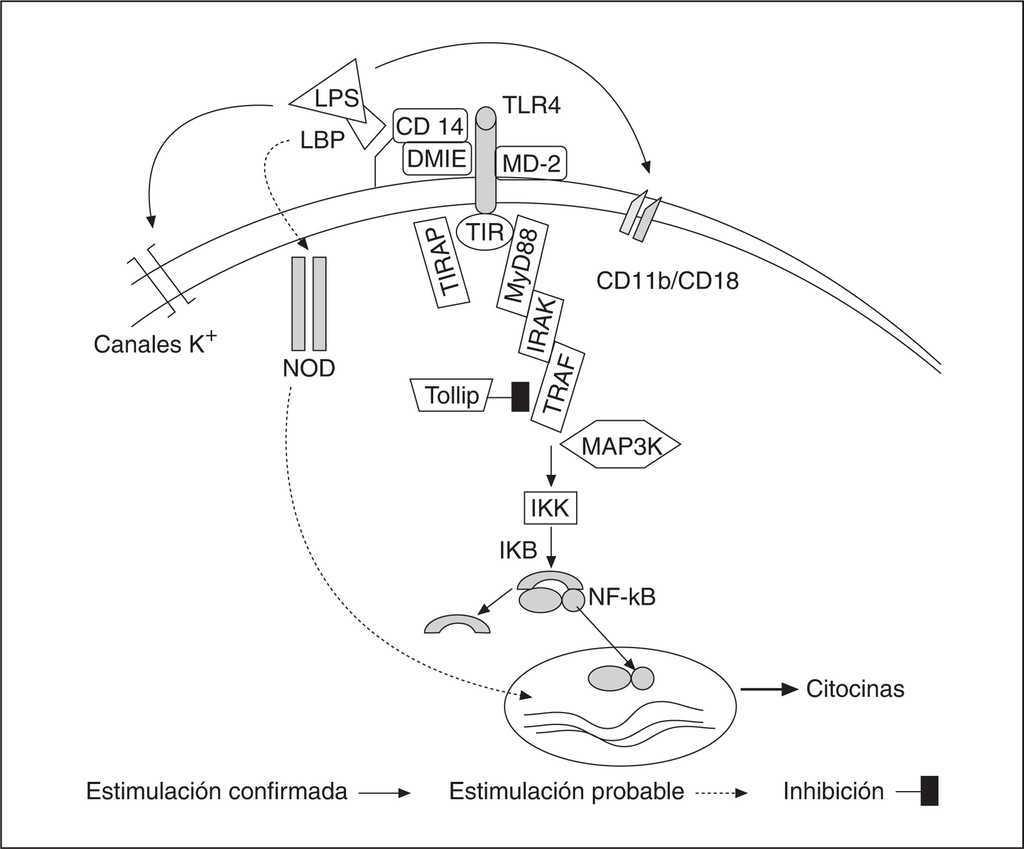
Figura 1. Actuación de la LPS sobre la membrana celular y señales celulares conocidas y probables desencadenadas por la misma. Las abreviaturas se encuentran en el texto.
Las células pueden también responder a la LPS por otra vía distinta a través de receptores intracelulares llamados proteínas NOD (nucleotide-binding oligomerization domain) que también presentan dominios ricos en leucina por los que interactúa con su ligando, el muramil dipéptido (NOD2) o el muramil tripéptido (NOD1), la unidad menor de peptidoglicano común a grampositivos y a gramnegativos. La expresión tanto de NOD1 como de NOD2 genera una respuesta de la LPS pero no del ácido lipoteicoico.
SEPSIS POR GRAMPOSITIVOS
La sepsis debida a grampositivos puede desencadenarse por dos mecanismos al menos, por producción de exotoxinas que actúan como superantígenos, o también a partir de componentes de la membrana celular que actúan como desencadenantes (peptidoglicanos, ácido lipoteicoico, lipoproteínas, modulina soluble en fenol). Estos mediadores interactúan en la membrana celular con el TLR2 y son menos activos que la LPS considerándolos a igual peso. No obstante, no existen aún trabajos clínicos convincentes que demuestren su presencia a concentraciones similares a las que se encuentran en los estudios experimentales.
Por lo que respecta a los superantígenos (fig. 2), éstos son moléculas que se unen a las células presentadoras de antígeno que participan en el MHC-II (complejo mayor de histocompatibilidad clase II), y también a las cadenas Vβ de los receptores de células T, desencadenando una producción masiva de citocinas proinflamatorias. Ejemplos reconocidos son las exotoxinas del Staphylococcus y del Streptococcus que producen el síndrome de shock tóxico. Además muestran, dependiendo de la secuencia del terminal amino de su dominio, afinidades para diferentes alelos HLA, de esa manera el superantígeno SPEA (streptococal pyrogenic exotoxin A) muestra mayor afinidad por el HLA-DQ que por el HLA-DR, lo que explicaría para algunos la selectividad tan marcada de los síndromes de shock tóxico. Otro hecho interesante es la hipersensibilidad que se produce a la LPS tras una agresión por superantígenos que justificaría el proceder de plantear estrategias frente al LPS aunque la sepsis sea producida por grampositivos.
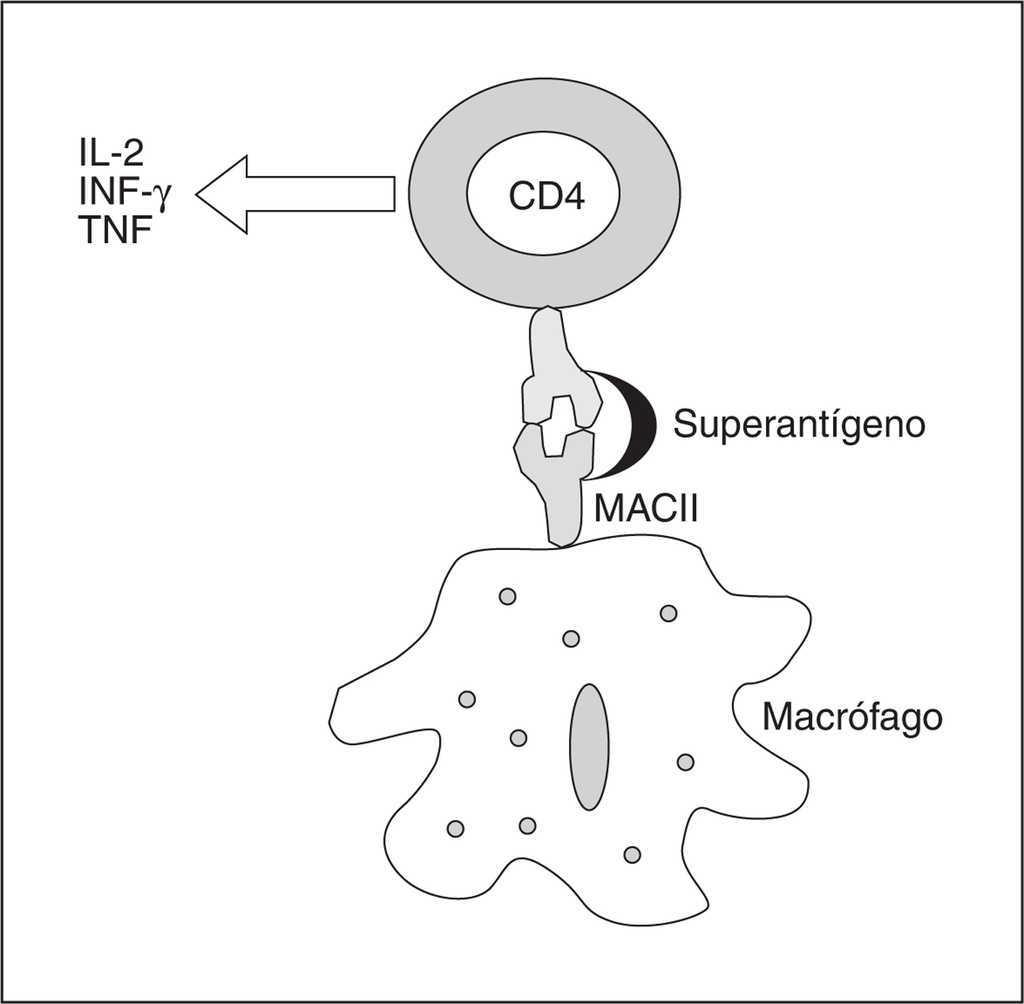
Figura 2. Esquema de la actuación de un superantígeno en una sepsis por grampositivos. Las abreviaturas se explican en el texto.
TOLL-LIKE RECEPTORS
Son proteínas transmembrana tipo I con un dominio extracelular rico en leucina y un dominio intracelular homólogo al receptor de interleucina 1 (IL-1). En el genoma humano se han clonado hasta ahora 10 moléculas que interactúan con partículas o componentes bacterianos conocidos como PAMP que comparten muchos patógenos. Los más conocidos son el TLR4 que es un ligando para la endotoxina, TLR2 ya citado para componentes de la membrana celular de los grampositivos, TLR5 cuyo ligando es la flagelina de las bacterias flageladas, TLR3 cuyo ligando es el ARN de doble cadena de los virus, TLR9 que recibe al ADN bacteriano con secuencias no metiladas CpG para la estimulación celular, y TLR1 que comparte con el TLR2 el ligarse a factores solubles procedentes de la Neisseria meningiditis. Por tanto, hasta el momento se considera que existe una señal común mediada por TLR9, TLR5 y TLR6; y señales específicas (TLR4 para LPS y TLR2 para ácido lipoteicoico)2.
TREM (TRIGGERING RECEPTOR EXPRESSED ON MYELOID CELLS)
Es una proteína de membrana que se ha identificado recientemente y que también está implicada en el reconocimiento bacteriano y de sus productos. Su ligando es desconocido. Su bloqueo en estudios experimentales hace que los monocitos estimulados con LPS expresen más citocinas proinflamatorias, y a la inversa, la adición de una proteína de fusión con TREM1 disminuye dicha producción, por lo que teóricamente puede convertirse en un futuro objetivo terapéutico.
MIF (MACROPHAGE MIGRATION INHIBITORY FACTOR)
Es una citocina que es expresada de una forma constitutiva por células epiteliales del pulmón y riñón, inmunes (macrófagos, eosinófilos) y endocrinas (pituitarias) en grandes cantidades y liberada tras exposición a otras citocinas proinflamatorias o a productos bacterianos. Se le atribuye funciones de modulación de la respuesta inmune a través del TLR4 permitiendo la respuesta rápida de macrófagos. En situaciones de sepsis grave y shock séptico se han detectado altos niveles de MIF. Tras estudios experimentales se ha llegado a la conclusión de que si se libera en exceso durante una sepsis, tiene la capacidad potencial de poner en peligro la supervivencia. Estas características se han evaluado por si tratamientos anti-MIF pudieran tener lugar en la sepsis. La interacción MIF-corticoides es singular pues aunque la acción de estos últimos se ha considerado siempre como antiinflamatoria, niveles bajos de los mismos inducen expresión de MIF por los macrófagos; y una vez liberado, el MIF actúa como un agente proinflamatorio capaz de inducir shock. Esta interacción no acaba de explicarse en el contexto clínico donde el aporte de dosis bajas de corticoides protege en la sepsis grave y en el shock séptico.
HMGP 1 (HIGH MOBILITY GROUP
PROTEIN 1)
Es una proteína cromosomal no histona que existe bajo distintas formas: unida a membrana, citoplásmica y nuclear, y que participa estabilizando los nucleosomas, facilitando la transcripción de genes y modulando la actividad de los receptores hormonales esteroideos. Se considera un mediador tardío de la sepsis donde se elevan sus niveles y esas altas concentraciones se han asociado a la evolución negativa del paciente. Experimentalmente, su bloqueo con anticuerpos policlonales lleva a la protección de endotoxinemias letales y a protección de la lesión pulmonar aguda. Esto hace considerar la posibilidad de que se convierta en diana terapéutica.
CASCADA DE LA INFLAMACION
La actuación de los factores de transcripción hacen que se liberen citocinas proinflamatorias que actuarán sobre otras células sanguíneas (linfocitos T y B, células natural killer [NK] y en un fenómeno de autorregulación sobre el propio monocito/macrófago), sobre médula ósea y sobre órganos diana (sistema nervioso central, hígado, glándulas suprarrenales, sistema adiposo, músculos estriados y probablemente sobre el sistema nervioso periférico entre otros). El episodio final tiene lugar en el territorio de la microcirculación donde las células endoteliales son a su vez estimuladas por la unión LPS-LBP-CD14 y se expresan moléculas de adhesión (ICAM, ELAM) que atraen a los polimorfonucleares que inician el rodamiento para posteriormente fijarse a la pared e iniciar la diapédesis hacia el foco infeccioso, mientras que por otro lado se produce una mayor cantidad de óxido nítrico (NO) no sólo de forma constitutiva sino fundamentalmente a partir de la NO-sintetasa inducible de los monocitos y otras células y alteraciones en la vía intrínseca y extrínseca de la coagulación y de la fibrinolisis (antitrombina III [AT-III], factor tisular, trombomodulina, proteína C) que conducen al atrapamiento de plaquetas y bloqueo de capilares, junto a la liberación de sustratos lipídicos como son la sobreproducción de prostaglandinas, especialmente de la serie 2 y leucotrienos de la serie 4, además de producción del PAF (platelet activating factor). Este mecanismo inflamatorio desencadena los efectos clínicos conocidos de fiebre, escalofríos, trastornos de la conciencia entre otros, y el síndrome de respuesta inflamatoria sistémica (SIRS) que es modulado por el propio organismo por diferentes líneas de regulación:
-Bloqueo de la LPS por la proteína incrementadora de la permeabilidad (BPI).
-Inhibición de la tirosinquinasa y de la proteinquinasa C.
-Producción de receptores solubles al factor de necrosis tumoral (TNF).
-Producción de IL-1 ra.
-Producción de citocinas antiinflamatorias como son la IL-4, IL-10 y factor transformador de crecimiento beta (TGF-β).
El bloqueo del TNF-α, aun cuando sobre el esquema general de la sepsis parece una diana terapéutica clara, no se ha mostrado muy satisfactorio en diferentes estudios.
PAPEL DE LAS CITOCINAS EN LA SEPSIS
Las citocinas conforman un sistema de modulación de respuestas proinflamatorias y antiinflamatorias de tal forma que regulan la respuesta del huésped a la sepsis. Algunas de ellas pueden ser detectadas únicamente en situaciones de shock séptico, mientras que otras presentan concentraciones plasmáticas elevadas en sepsis o sepsis grave. Esto podría explicarse por el hecho de que, por ejemplo, el TNF, y la IL-1 α y β se piensa que son producidas por un mecanismo paracrino, por lo que sus niveles plasmáticos no se correlacionan de forma estrecha con su acción mientras que otras como la IL-6 tienen una actividad de mayor ámbito sistémico además de su ambivalencia al comportarse bien como mediador proinflamatorio o bien como antiinflamatorio dependiendo de cada situación.
Muchas de las citocinas tienen sus propios receptores individuales que están localizados en las membranas celulares y se unen al dominio extracelular de su receptor activando las tirosinquinasas intracelulares. Una vez que la señal se ha iniciado, la citocina con su componente extracelular del receptor se disocia y forma un complejo soluble que puede ser detectado en el plasma. La regulación de las citocinas puede hacerse bien por la modulación de su actividad a través de sus antagonistas plasmáticos o por un sistema de retroalimentación por los miembros distales de la vía de las citocinas sobre los miembros más proximales, tanto de forma directa como indirectamente a partir de hormonas.
Dado el desarrollo en el estudio de las citocinas actualmente se pueden detectar más de 50 de ellas en el plasma. No obstante, queda por determinar la función o funciones de buena parte de ellas así como la ambivalencia dependiendo de las condiciones del medio donde se está desarrollando el proceso inflamatorio.
RESPUESTA INFLAMATORIA
Constituye un mecanismo complejo y fundamental para el mantenimiento de la vida donde participan numerosas subcascadas y se implican diversos sistemas arriba citados y cuyos protagonistas más conocidos son fundamentalmente el TNF-α, IL-1, IL-6, interferón γ (IFN-γ), IL-18, además de las quimiocinas IL-8 y la familia MCP (monocyte chemotactic protein) que atraen a los polimorfonucleares y activan a los macrófagos.
El factor de necrosis tumoral, TNF como es universalmente reconocido en la extensa bibliografía al respecto, fue la primera citocina implicada en la patogénesis de la sepsis. Los niveles séricos de TNF se encuentran elevados en pacientes con sepsis aunque dicha elevación depende tanto de la gravedad como del tipo de proceso séptico. Mientras que en la meningococemia este paralelismo es evidente, no sucede lo mismo en las situaciones de sepsis abdominal donde no se puede establecer de forma clara una relación con la supervivencia. En esta discordancia se implican la presencia de receptores solubles circulantes y el momento de la determinación plasmática de TNF, dada su relativamente corta vida media.
Las diversas actuaciones intentando bloquear esta citocina no han dado resultados satisfactorios. En el estudio INTERSEPT3 donde se empleó un anticuerpo monoclonal (AcM) murino diseñado contra TNF-α recombinante sobre un total de 564 enfermos, de los que la gran mayoría (420) se encontraban en shock séptico, no se encontró ningún beneficio sobre la supervivencia a los 28 días frente al grupo control, aun cuando en la supervivencia más tardía se apreciaba en el grupo de tratamiento un retraso en la aparición del primer fallo de órganos y una recuperación más rápida de la situación de shock.
En el primero de los estudios del North American Sepsis Trial (NORASEPT I)4, sobre un total de 994 enfermos, de los que 478 estaban en situación de shock séptico, se empleó un AcM frente a TNF-α en un ensayo clínico aleatorizado, controlado, doble ciego, prospectivo y multicéntrico. Se establecieron tres grupos, dos recibieron diferentes infusiones del AcM y el tercero recibió un placebo, sin que se encontraran diferencias con respecto a la mortalidad entre los grupos de tratamiento y el grupo placebo. En el subgrupo de pacientes con shock se encontró una tendencia hacia la disminución de la mortalidad global en el grupo de tratamiento frente al grupo control, que se ponía más de manifiesto tres días tras la infusión pero sin diferencias entre las distintas dosis administradas de AcM, donde incluso la reducción de la mortalidad era más acentuada entre los pacientes tratados con dosis más bajas. Esta tendencia no se mantenía al estudiar la mortalidad en estos enfermos con shock a los 28 días.
Un nuevo estudio, el NORASEPT II, se realizó empleando el mismo AcM del INTERSEPT en 1.879 enfermos en shock séptico5 a las dosis bajas que mostraron mejor comportamiento en el NORASEPT I (7,5 mg/kg), pero no se encontró mejoría en la reversión del shock ni en la prevención del mismo en ninguno de los subgrupos establecidos de acuerdo a las mayores concentraciones de IL-6 o TNF plasmáticos.
En las estrategias dirigidas al empleo del bloqueo del TNF a partir de los receptores solubles p55 y p75 los resultados son dispares. En un estudio empleando un combinado IgG-1 humano y p75 (porción extramembrana del receptor soluble tipo II del TNF) se sugería que a dosis más elevadas se incrementaba la mortalidad, invocando un posible mecanismo de liberación del TNF en un segundo tiempo tras un bloqueo inicial6,7.
Con respecto al receptor p55, en un trabajo sobre pacientes con sepsis con y sin shock, y a diferentes dosis frente a placebo, no se encontró un descenso significativo de la mortalidad, aunque en el subgrupo de pacientes más graves, aquellos que recibían la dosis más elevada mostraban una tendencia hacia una reducción de la mortalidad al día 288. Un nuevo estudio multicéntrico muy amplio no pudo probar posteriormente esta hipótesis9.
La IL-1 es otra citocina proinflamatoria de notable actividad en la sepsis. Engloba a una familia de mediadores que comprende por un lado a dos formas con distinto peso molecular, IL-1 alfa e IL-1 beta, siendo la segunda la más estudiada. Asimismo ambas presentan dos tipos de receptores (I y II) de cuya unión se induce una señal de transducción que induce la activación de factores de transcripción tales como el NF-κB y la proteína 1 de activación (AP-1). Pero además la familia cuenta con antagonistas a receptores de IL-1 (IL-1ra) que son inhibidores solubles circulantes que se unen a los receptores y que se liberan siempre tras la elevación plasmática de IL-1, por lo que como en el caso del TNF constituirían un mecanismo de autocontrol en la respuesta inflamatoria. La elevación encontrada en la sepsis se correlaciona de forma positiva con la evolución pero el hecho de que la administración exógena fracasa en su objetivo de mejorar la evolución hace que se haga dudar de su verdadera capacidad para constituir mediadores de resolución de la sepsis. En un estudio prospectivo, aleatorizado, doble ciego, controlado por placebo y multicéntrico, sobre 696 pacientes con sepsis grave y shock séptico realizado en 91 hospitales de Europa y Norteamérica, la administración de 100 mg de IL-1ra recombinante humana en bolo seguida de una perfusión de 2 mg/kg/h durante 72 horas frente a tratamiento con placebo, no se pudo demostrar una reducción estadísticamente significativa de la mortalidad10. La IL-1 es de muy difícil detección en los pacientes con sepsis pero cuando se detecta, presenta una relación clara con la mortalidad11 y su papel de mediador es muy similar al del TNF actuando en ocasiones de forma coordinada compartiendo órganos diana mientras que en otras actúa de manera independiente.
La IL-6 es un mediador con actividad ambivalente, considerado por unos durante mucho tiempo como citocina proinflamatoria, y clasificado por otros como antiinflamatoria, aunque probablemente comparta ambas actividades o bien, como piensan otros12, no sea un mediador directo sino un marcador de inflamación sistémica y que refleje solamente los efectos netos de la producción previa de TNF e IL-1, o bien que la IL-6 sea pro-inflamatoria sólo en presencia de otras citocinas. La infusión de IL-6 provoca fiebre pero no produce los efectos hemodinámicos de las otras citocinas referidas. La IL-6 es la citocina cuyos niveles séricos al ingreso están más estrechamente relacionados con la muerte, aunque dichos niveles descienden rápidamente a pesar de que persista la sepsis y el fallo de órganos. Sin embargo, en diferentes estudios con actuación terapéutica sobre la sepsis, este descenso fue más rápido que en los distintos grupos controles, aunque eso no significó siempre mejor evolución pues en el tercero de ellos no hubo mejoría evolutiva13.
Y aunque se sugiere la posibilidad de que el descenso de la IL-6 se asocie con la resolución de la sepsis, esto no queda claro porque el descenso antes citado tras las primeras 24-48 horas se encuentra tanto en pacientes que sobreviven como en los que fallecen, aunque en un estudio se notificó que la IL-6 permanecía elevada en los pacientes que no sobreviven al décimo y décimotercer días14.
La IL-8 es un péptido que junto con una superfamilia de 10 ó más citocinas proinflamatorias se ha designado como quimiocinas. La IL-8 produce la estimulación de la quimiotaxis, aumento de la liberación de enzimas lisosomales y sobrerregulación de receptores del complemento. La infusión de IL-8 induce granulocitopenia inicial seguida de granulocitosis. Asimismo, cuenta con dos receptores de alta afinidad (I y II) siendo el II compartido por otros mediadores, la señal de transducción se efectúa a partir de la movilización del calcio intracelular y activación de la proteinquinasa C. La IL-8 se detecta en el suero de pacientes con sepsis aunque solamente y por ahora en pocos trabajos se ha correlacionado con la mortalidad.
RESPUESTA ANTIINFLAMATORIA
Frente a la cascada proinflamatoria, se produce un efecto de contrarregulación para modular dicha cascada que se basa en la producción de antagonistas de los receptores solubles para el TNF e IL-1, en receptores-señuelo de la IL-1, en inactivadores del complemento y en producción de citocinas claramente antiinflamatorias cuyo paradigma es la IL-10, el TGF-ß o la IL-4. Esta respuesta antiinflamatoria vendrá mediada por la supremacía que se establezca en la diferenciación de los linfocitos Th0 en Th1 (proinflamatorios) o Th2 (antiinflamatorios), esta diferenciación está mediada por diversos factores tales como el tipo de patógeno, el tamaño del inóculo y el lugar de la infección, pero entre ellos la presencia de citocinas dominantes puede determinar el proceso (IL-2 para Th1 y IL-4 para Th2).
La IL-10 es una citocina antiinflamatoria. Entre sus actividades se encuentra la supresión de IFN-γ por parte de los linfocitos T cooperadores y las células NK, la supresión de citocinas proinflamatorias (TNF, IL-1, IL-6 e IL-8) por los monocitos, al mismo tiempo que sobrerregula la producción de IL-1 ra. De forma inversa, la inhibición de IL-10 produce incremento de los niveles circulantes de TNF e IL-6. Los enfermos con shock séptico presentan niveles elevados de IL-10 que son independientes de la presencia o no de infección documentada microbiológicamente pero sí están relacionados con la presencia de bacteriemia y con la gravedad del shock séptico medida por la escala APACHE II.
Aunque tanto la IL-10 como el TGF-ß actuarían como factores que resuelven una situación de sepsis, esta actuación puede desembocar en un estado de relativa inmunoparesia que puede predisponer a los pacientes a infecciones nosocomiales. En pacientes con sepsis se han encontrado niveles circulatorios elevados de IL-10, aunque no en todos los pacientes (al contrario del TGF-ß cuyas concentraciones séricas suelen estar bajas o normales en la sepsis). Estos niveles se hacen máximos a las 48 h y permanecen durante 3-5 días, y están en relación con la evolución y con las concentraciones de citocinas proinflamatorias. A partir de estudios animales se considera que aunque la IL-10 pueda desempeñar un papel importante en la resolución de la sepsis, el tiempo de administración es crítico.
Dentro de la respuesta antiinflamatoria se alinean mediadores como los IL-1ra (antagonistas de receptores de IL-1) antes referidos, y receptores como el tipo A (SR-A).
APOPTOSIS EN LA SEPSIS
Dentro de los mecanismos contrarreguladores se halla la apoptosis. Esta apoptosis que se produce en la sepsis en los linfocitos B, células T CD4, células dendríticas y células epiteliales gastrointestinales15 lleva a una inducción de anergia o de producción de citocinas antiinflamatorias que dificultan o disminuyen la respuesta al patógeno, ya que se produce, por lo que respecta a linfocitos y células dendríticas, cuando cabría esperar la expansión clonal de los mismos y ello lleva a un descenso en la producción de anticuerpos (linfocitos B), en la activación de los macrófagos (CD4) y en la presentación de antígenos (células dendríticas). Inversamente, la necrosis celular estimula la respuesta inmune y aumenta las defensas frente a los gérmenes. Por lo que respecta a los mecanismos de producción de apoptosis, ésta puede ser inducida por falta de estimulación antiapoptótica que lleva a una pérdida de la estructura de la mitocondria y liberación de citocromo C y estimulación del factor APAF-1 (apoptotic protease-activating factor), o por ligandos que se unen a los receptores de la familia TNF tales como el TRAIL (TNF-Related Apoptosis- Inducing Ligand) y el FAS-L. Ambos mecanismos desencadenan la cascada de las caspasas que conlleva la degradación de los materiales genéticos y las proteínas celulares estructurales, formándose cuerpos apoptóticos que son destruidos por los fagocitos circundantes. Probablemente, la apoptosis de linfocitos se deba a la liberación endógena de corticoides inducidos por la agresión.
CASCADA DEL COMPLEMENTO
En la sepsis, tanto la LPS como inmunocomplejos circulantes, reactantes de fase aguda o receptores como el ya referido lectina unida a manano, inducen activación de ambos sistemas del complemento. El objetivo es el ataque a las membranas del complejo C5b-9 formando poros y propiciando su destrucción. El C5a tiene distintas acciones con trascendencia proinflamatoria. En la sepsis, el C5a se encuentra más elevado cuanto más grave es el cuadro séptico y esa elevación se relaciona de forma directa con la supervivencia y el fallo de órganos. La producción de C5a en la sepsis en seres humanos se asocia a un efecto procoagulante y a una alteración de la génesis de citocinas y de la actividad sobre la producción de aniones superóxidos por los neutrófilos, liberación de enzimas granulares por los fagocitos y efectos de vasodilatación y aumento de la permeabilidad por lo que se ha planteado una línea de tratamiento bloqueándolo. En estudios animales estos trabajos han mostrado una mejoría de la supervivencia. Queda pues constatar estos resultados en la clínica.
Los conocimientos actuales sobre la fisiopatología de la sepsis hacen que, ante el abrumador aporte de datos provenientes de la investigación básica y experimental, la aplicabilidad de los mismos a la clínica requieran una meditación prudente dada la dificultad en la identificación de cuál o cuáles puedan ser los mecanismos o los sustratos principales que produzcan una modificación sostenida y reproducible en la respuesta inflamatoria y lo que es más importante en su trascendencia sobre la supervivencia de la sepsis. La experiencia previa bloqueando diferentes mediadores no ha llevado más que a la insatisfacción y a la decepción. El descubrimiento de posibles nuevos objetivos terapéuticos tales como la HMGP-1, el MIF, el C5a, alienta, no obstante, expectativas diferentes para su introducción en la clínica en un futuro próximo.

