







Editado por: F. Baigorri-González y J.º. Lorente Balanza
Última actualización: Abril 2005
Más datosINTRODUCCIÓN
Durante muchos años el endotelio fue considerado una barrera inerte que separaba el torrente circulatorio de los tejidos subyacentes. Los avances en la comprensión de los procesos involucrados en la respuesta del huésped a la lesión han permitido reconocer a esta población celular como un órgano verdadero que ocupa un lugar fundamental en el montaje de dicha respuesta.
Ampliamente distribuidas en el cuerpo humano, las células endoteliales tapizan el compartimiento vascular que alcanza una superficie de 4.000 a 7.000 m2 y cumplen múltiples funciones donde destacan: regulación del tono vasomotor, balance local de mediadores pro y antiinflamatorios, regulación del desplazamiento de células y nutrientes, mantenimiento de la fluidez sanguínea y generación de neovasos1.
En la sepsis se produce una activación endotelial masiva, exagerada y sostenida que se caracteriza por desequilibrio entre mediadores pro y antiinflamatorios y pérdida del tono vasomotor. En este caso la respuesta es "desadaptada" (disfunción endotelial) y puede acabar con la vida del individuo afectado. El término disfunción endotelial se relaciona con ambos tipos de respuesta (local o sistémica) y se refiere principalmente a aquellos casos en que la respuesta endotelial implica, de una u otra manera, un coste para el huésped2.
En este capítulo nos referiremos a 4 aspectos de la activación endotelial: a) activación de la célula endotelial como respuesta adaptativa normal; b) disfunción endotelial en la sepsis; c) daño endotelial e insulto hipóxico/isquémico, y d) endotelio y regulación del tono vasomotor (unidad reguladora mioendotelial).
ACTIVACION DE LA CÉLULA ENDOTELIAL COMO RESPUESTA ADAPTATIVA NORMAL
En condiciones normales las células endoteliales se encuentran continuamente reconociendo y respondiendo en forma activa frente a los cambios que ocurren en el ambiente extracelular local, como sucede en presencia de bacteremia, trauma, isquemia- reperfusión, etc. En otras palabras, la activación de la célula endotelial se produce como respuesta adaptativa normal y la forma de presentación y duración de ésta dependerá del tipo de estímulo, sitio donde se desarrolla y momento en que ocurre su activación dentro del sistema circulatorio. Este concepto temporoespacial de la activación endotelial se ha denominado "heterogenicidad endotelial" o "diversidad vascular". Cabe destacar además que la respuesta endotelial no es un fenómeno del "todo o nada" (on-off), sino que representa un espectro amplio que depende del estado de salud o enfermedad del huésped1.
Así, cuando la respuesta endotelial beneficia al organismo en cuestión, ésta puede ser considerada adaptativa y fisiológica, como ocurre en la invasión local por un patógeno. En este caso, la liberación de mediadores inflamatorios, marginación de leucocitos y activación de la coagulación cumplen la función de rodear la infección e impedir que ésta se distribuya ampliamente en el resto del organismo.
Transducción de la señal inducida por lipopolisacáridos
El lipopolisacárido (LPS) es el responsable de la iniciación de la cascada inflamatoria en las infecciones producidas por bacterias gramnegativas. Su actividad biológica es mediada por una glucoproteína de fase aguda de síntesis hepática que posee alta afinidad para LPS, denominada proteína de enlace de LPS (LBP). La importancia de esta última en la activación de la sepsis ha quedado demostrada por la menor sensibilidad a LPS observada en ratas carentes del gen de LBP.
La LBP forma un complejo con LPS (LPS-LBP) y funciona como una proteína de transferencia que acarrea LPS hacia receptores ubicados en la superficie celular. El complejo LPS-LBP ejerce su actividad al entregar LPS a un receptor de membrana específico denominado mCD14 que se encuentra en la superficie de macrófagos, monocitos y neutrófilos.
Existen resultados contradictorios con respecto a la expresión de este receptor en la superficie de las células endoteliales; sin embargo, algunos autores han logrado estimular células endoteliales mCD14 negativas. En ausencia de mCD14, LPS puede actuar sobre las células endoteliales por medio de la forma soluble de este receptor (sCD14). Este último es liberado a la circulación desde los monocitos activados y es capaz de actuar como mediador de la actividad de LPS en una forma LBP-dependiente similar a mCD143.
Ambas formas de CD14 se comportan como moléculas de reconocimiento que interactúan no sólo con LPS, sino también con otros componentes microbianos como peptidoglicano soluble y ácido lipoteicoico.
El mCD14, al carecer de dominio intracelular, es incapaz de iniciar por sí mismo una respuesta celular, razón por la cual debe acompañarse de proteínas transmembrana. Entre éstas destacan los receptores toll-like (TLR) identificados como indispensables para inducir la activación de mediadores intracelulares tales como proteínas quinasas y factor nuclear kappa-B (NF-κB).
La célula endotelial expresa predominantemente dos tipos de TLR, TLR-4 y TLR-2. Ambos son regulados por factores relacionados con la inflamación, entre ellos LPS, factor de necrosis tumoral-α (TNF-α) e interferón-γ (IFNγ).
En los mamíferos, el TLR-4 es probablemente la proteína transmembrana más importante involucrada en la transmisión de la señal inducida por LPS; no obstante, es necesario mencionar que se han identificado otros tipos de proteínas como el fMLP, que se comportan de manera similar.
Recientemente en una variedad de ratas resistentes a LPS (C3H/HeJ y C57BL/10ScCr) se demostró que la mutación del gen para TLR-4 les confería resistencia a los efectos de la endotoxina bacteriana. Sorprendentemente, esta anomalía no pudo ser corregida por transfección de sus líneas celulares con receptores TLR-4 lo que hizo plantear la existencia de moléculas adicionales.
TLR-4 para ejercer su acción requiere la presencia de una glucoproteína accesoria denominada MD-2, ésta se une al dominio extracelular de TLR-4 otorgándole mayor capacidad de respuesta frente a LPS. Probablemente cada miembro de la familia de TLR necesita una proteína accesoria específica para reconocer los diferentes componentes bacterianos3.
Factor de transcripción NF-κB
Los efectos proinflamatorios del LPS se cree que están mediados por activación del NF-κB que se encuentra presente tanto en células mieloides (monocito-macrófago, polimorfonucleares [PMN]) como en células endoteliales. Este factor desempeña un papel central en la regulación de la transcripción de citocinas, moléculas de adhesión y otros mediadores que dependerán del tipo de célula estimulada.
El NF-κB forma parte de una familia de proteínas estructuralmente relacionadas que contienen una región aminoterminal denominada dominio homólogo Rel (RDH), donde se encuentra el dominio de dimerización y la señal de localización nuclear (NLS).
Cuando la célula se encuentra en estado quiescente, en el citoplasma celular NF-κB, heterodímero constituido por las subunidades p50 (NF-κB1) y p65 (RelA), se encuentra unido a una proteína inhibidora conocida como IκB que enmascara la NLS4.
Mediadores intracelulares y activación del NF-κB
Como se mencionó, el NF-κB unido al IκB permanece en el citoplasma como un factor de transcripción latente. La liberación del NF-κB se consigue a través de la fosforilación y degradación proteolítica del IκB en el citoplasma, proceso que puede ocurrir en respuesta a una amplia gama de estímulos tales como TNF-α, interleucina 1B (IL-1ß), LPS, ARN doble hebra, especies reactivas de oxígeno (ERO), radiación gamma, etc.
El IκB está constituido por dos subunidades inhibidoras (IκBα e IκBß). Ambas son susceptibles de ser fosforiladas y esto ocurre a nivel de residuos de serina específicos ubicados en su extremo aminoterminal (Ser32, Ser36). Este proceso se inicia con la activación de dos quinasas de IκB (IKKα e IKKß), también conocidas como IKK-1 e IKK-2, en respuesta a los estímulos antes citados.
Luego, las quinasas de IκB forman homo y heterodímeros que se unen a una tercera proteína conocida como modulador esencial de NF-κB (NEMO). Este complejo formado por IKK-1, IKK-2 y NEMO pasa a formar parte, junto a otras proteínas, de una estructura de alto peso molecular denominada IKK signalosoma, capaz de acoplarse a otras quinasas activadoras corriente arriba, entre ellas, proteínas quinasa-quinasa-quinasa activadas por mitógenos (MAP3K), quinasa inductora de NF-κB (NIK) y quinasa quinasa-1 reguladora de la señal extracelular (MEKK-1).
A su vez, para que estas quinasas corriente arriba continúen con el proceso de fosforilación de IκB, requieren interactuar directamente con diferentes factores que se encuentran asociados a los receptores de TNF-α e IL-1. Estos factores forman parte de complejos de señal citoplasmática receptor-específicos, entre los que destacan TRAF1, TRAF2, receptor de TNF 1-asociado al dominio proteico de muerte (TRADD) y TRAF6 donde TRADD se encuentra asociado con receptores de TNF-α y TRAF6 con los de IL-15.
Un ejemplo de ello, es la capacidad que tiene NIK para unirse directamente con TRAF2 e iniciar la activación del NF-κB. Aunque no existe consenso, parece que la sobreexpresión de NIK y MEKK-1 conducen a la fosforilación de IKK-1 e IKK-2, donde NIK expresa preferencia por IKK-1 y MEKK-1 por IKK-24.
Una vez concluida la fosforilación de IκB, éste es ubiquitinado por la ubiquitina ligasa E3, miembro terminal de una cascada de enzimas conjugantes de ubiquitina, para luego ser degradado por el proteasoma 26S. En ese momento el heterodímero p50-p65 libre es translocado al núcleo, donde se une a secuencias κB en regiones promotoras de genes específicos que codifican mediadores activados transcripcionalmente por este factor. En el caso de las células endoteliales los principales mediadores serán moléculas de adhesión, tales como E-selectina, moléculas de adhesión intracelular (ICAM-1) y molécula de adhesión intercelular (VCAM-1)4 (fig. 1).
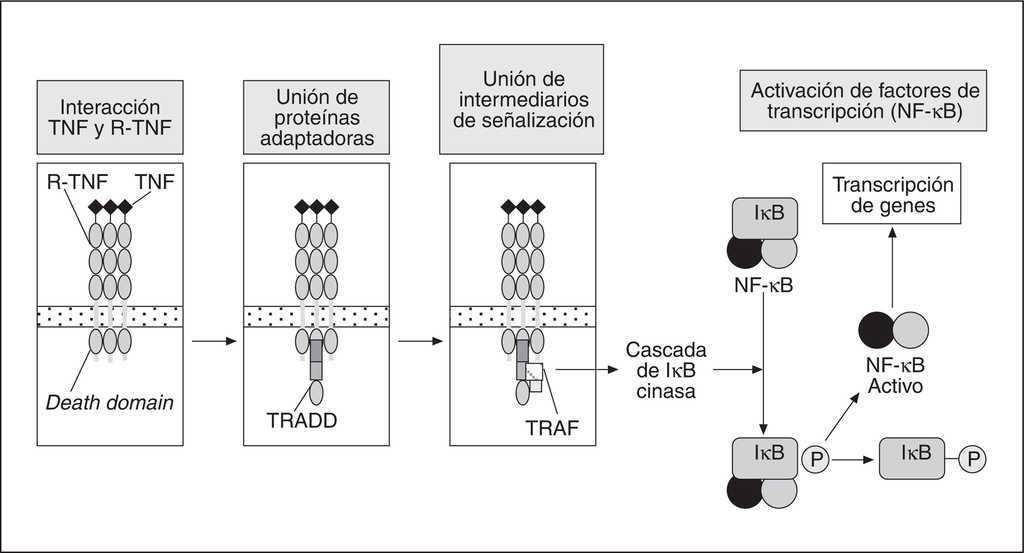
Figura 1. Mediadores intracelulares y activación del factor nuclear kappa-B (NF-κB). TNF: factor de necrosis tumoral; TRADD: receptor de TNF1 asociado al dominio proteico de muerte.
DISFUNCION ENDOTELIAL EN LA SEPSIS
El compromiso de la función endotelial forma parte de la respuesta multicelular integrada que el huésped es capaz de montar frente a un patógeno agresor. Esta disfunción puede culminar en una alteración grave de la homeostasis vascular que se caracteriza por oclusión microvascular de magnitud variable que puede llevar a un desequilibrio entre la oferta y demanda de oxígeno a nivel tisular6.
En la sepsis, la célula endotelial cambia desde un fenotipo quiescente (anticoagulante, antiadhesivo, vasodilatador) a uno activado (procoagulante, proadhesivo, vasoconstrictor) donde ocurren cambios estructurales y funcionales que la caracterizan (tabla 1). Tanto las células endoteliales como los monocitos participan en la activación paralela de las cascadas de inflamación y coagulación que tendrán como fin eliminar el agente patógeno. Sin embargo, simultáneamente esta respuesta puede generar daño colateral en tejidos normales que puede contribuir al desarrollo de disfunciones de órganos cuya consecuencia será un aumento de la morbimortalidad en este tipo de pacientes7.

En condiciones normales, la actividad biológica de los mediadores involucrados en la respuesta inflamatoria está sometida a un estricto control por parte de inhibidores específicos. En el paciente séptico, en cambio, este balance se encuentra deteriorado y se manifiesta a través de cambios considerables en las tasas de producción de los distintos mediadores con el desarrollo de un estado de desequilibrio entre pro y antiinflamación8 (fig. 2).
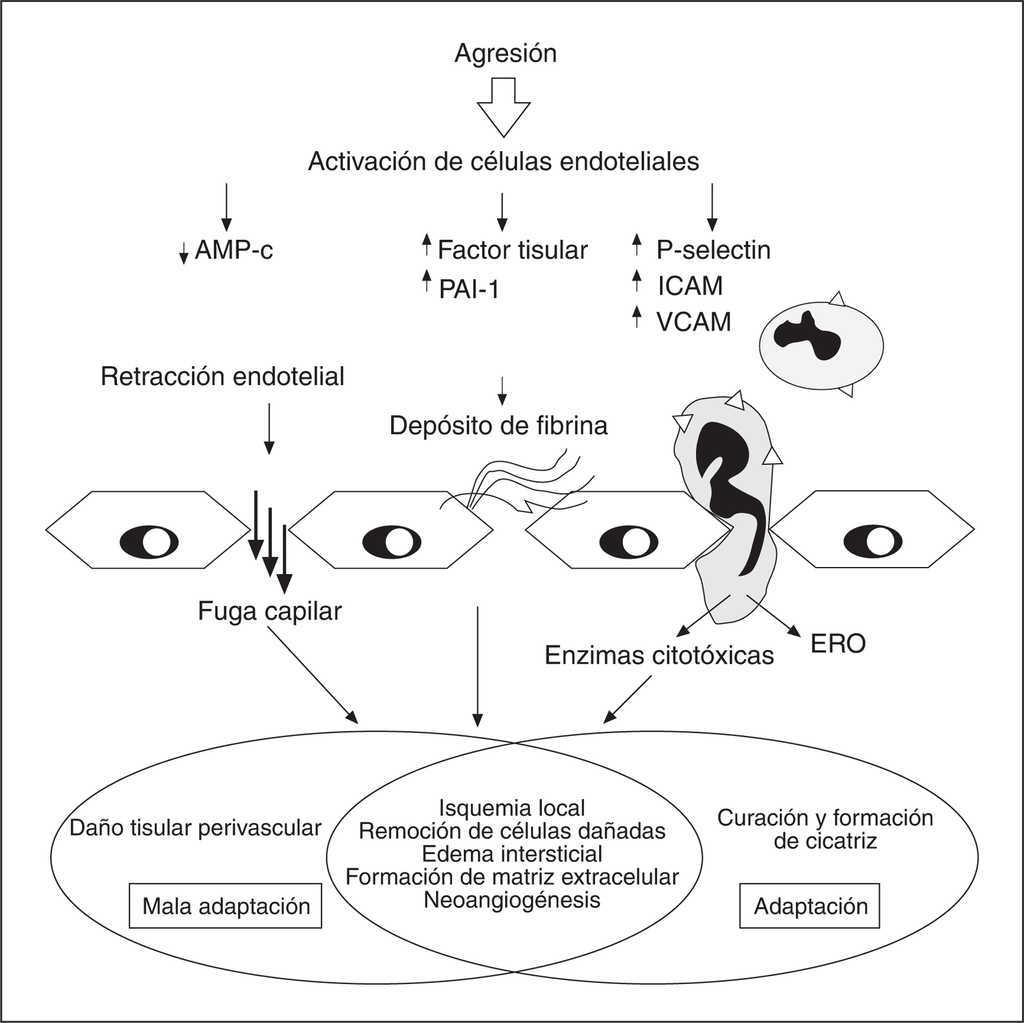
Figura 2. Respuesta endotelial a la agresión. Modificada de Ten V, Pinsky D13. AMPc: 3', 5'-adenosin monofosfato cíclico; PAI-1: inhibidor del activador del plasminógeno 1; ICAM: moléculas de adhesión intracelular; VCAM: moléculas de adhesión intercelular; ERO: especies reactivas de oxígeno.
A continuación haremos una breve descripción de las características más importantes que adquiere la célula endotelial en su estado activado: a) propiedades procoagulantes; b) propiedades proadhesivas; c) pérdida de la función de barrera, y d) apoptosis de la célula endotelial
Propiedades procoagulantes
Los mediadores inflamatorios liberados durante la respuesta inflamatoria pueden interactuar con las células endoteliales e inducir en ellas un fenotipo procoagulante. Estudios in vitro han demostrado que estas células disminuyen la síntesis de trombomodulina, activador del plasminógeno tisular y heparán sulfato. Al mismo tiempo ocurre un aumento de la expresión del factor tisular, del inhibidor del activador del plasminógeno 1 (PAI-1) e incremento en la generación de micropartículas fosfolipídicas procoagulantes que contribuyen al daño celular.
El aumento en la expresión del factor tisular en la superficie de monocitos circulantes y macrófagos tisulares lleva a la activación de la vía extrínseca de la cascada de coagulación, generación de trombina, formación de fibrina y su posterior amplificación por la vía intrínseca de la coagulación. La activación de esta última incrementa la producción de citocinas y quimioquinas por parte de las células endoteliales y aumenta la expresión de moléculas de adhesión. Se ha propuesto además que la fibrina, al inmovilizar los patógenos en la superficie de los leucocitos, facilitaría su fagocitosis.
Factores hemodinámicos, tales como la reducción del gasto cardíaco y la vasoconstricción asociados a lesiones vasculares oclusivas, pueden favorecer la formación local de coágulos debido a una reducción en la remoción de serinas proteasas activadas. En estas circunstancias la respuesta inflamatoria se amplifica, ya que estas enzimas son responsables de activar sustratos corriente abajo en la cascada de la coagulación e interactuar con receptores de membrana de monocitos y células endoteliales.
Se ha demostrado que junto con la activación de la vía extrínseca de la coagulación ocurre una marcada disminución de la actividad anticoagulante natural (proteína C, antitrombina III y trombomodulina) favoreciendo el perfil procoagulante del estado séptico. Estudios clínicos han puesto en evidencia que la activación de la coagulación, el depósito difuso de fibrina intravascular y la inhibición de la fibrinólisis estarían relacionados también con el desarrollo de disfunción de órganos y con aumento de la mortalidad. Además, hay datos consistentes para afirmar que las propiedades procoagulantes descritas durante la sepsis no son homogéneas en los distintos órganos y tejidos analizados, planteando así que la respuesta del endotelio en los diferentes lechos vasculares está sometida a un tipo especial de regulación9,10.
Propiedades proadhesivas
Frente a los mediadores inflamatorios el endotelio responde expresando en su superficie moléculas de adhesión tales como P-selectina (CD62P), E-selectina (CD62E), ICAM-1 (CD54), VCAM-1 (CD106), PECAM-1 y CD99. Todas ellas en conjunto favorecerán el tráfico de leucocitos desde el torrente circulatorio al tejido subyacente, principalmente en el territorio precapilar y en los capilares de órganos nobles como corazón y pulmón. Además, el endotelio aumenta el reclutamiento de plaquetas en la pared de los vasos a través de un mecanismo dependiente de GPIIb/IIIa. Cabe destacar que ninguna de estas observaciones ocurre de manera generalizada, sino más bien como eventos localizados en ciertos órganos y/o segmentos vasculares3 (fig. 3).

Figura 3. Sepsis y tráfico de leucocitos. Modificada de Peters K, et al3. LPS: lipopolisacárido; LBP: proteína de enlace de LPS; TLR: receptores toll-like; NF-κB: factor kappa B; IL-1B: interleucina 1B; TNF-α: factor de necrosis tumoral alfa; ICAM-1: moléculas de adhesión intracelular; VCAM-1: moléculas de adhesión intercelular.
Pérdida de la función de barrera
En la sepsis la redistribución de fluidos al espacio extravascular se ve favorecida por el incremento de la permeabilidad endotelial o pérdida de la función de barrera. Datos experimentales sugieren que este aumento de la permeabilidad es mediado por LPS y se relaciona con la escisión enzimática de proteínas que forman parte de las uniones intercelulares, dando como resultado daño estructural de la célula endotelial. Estudios in vivo e in vitro han demostrado que el TNF-α es uno de los responsables de este trastorno y que esta actividad podría verse potenciada por trombina3.
Apoptosis de las células endoteliales
En condiciones normales un porcentaje muy bajo de las células endoteliales (< 0,1%) experimenta muerte celular programada (apoptosis) sugiriendo que éste es un proceso con alto grado de regulación. En forma experimental se ha demostrado que tanto agentes patógenos, endotoxina, citocinas proinflamatorias, ERO y condiciones de isquemia/reperfusión son capaces de inducir apoptosis a través de up-regulation de proteínas claves en dicho proceso tales como el homólogo de Bcl-2, A1, A20 y proteínas con dedos de zinc.
Las células endoteliales en apoptosis aumentan la respuesta inflamatoria al favorecer la acción paracrina de citocinas proinflamatorias sobre la inducción de ICAM-1, VCAM-1, ERO, actividad procoagulante y activación del complemento.
La importancia de la muerte celular por apoptosis, que sufren tanto células endoteliales como linfocitos y células epiteliales del tubo digestivo, ocupa un lugar central en la patogenia de la sepsis severa/ shock séptico y ha sido demostrada en necropsias de pacientes fallecidos por disfunción multiorgánica11.
DAÑO ENDOTELIAL E INSULTO HIPOXICO/ISQUÉMICO
La presencia de oxígeno es obligatoria para la supervivencia de la célula eucariota, ya que la respiración mitocondrial es más eficiente que la glucólisis anaeróbica en la generación de energía a partir de glucosa. Las células endoteliales, comparadas con otros tipos celulares como miocardiocitos, neuronas y células del túbulo renal, son más resistentes a la disminución del contenido de oxígeno en el medio, tolerancia que posiblemente proviene de la exposición frecuente de éstas a niveles reducidos de oxígeno como sucede en la arteria pulmonar y tejidos en cicatrización. Al parecer, esta resistencia a la hipoxia tiene relación además con la activación de proteínas antiapoptóticas que van en paralelo con la magnitud del insulto más que con la naturaleza de éste.
La respuesta de la célula endotelial a la hipoxia involucra mecanismos dirigidos a conservar la energía como down-regulation de las funciones que poseen altos requerimientos energéticos (transporte de iones) e implementación de vías alternativas para la síntesis de adenosina trifosfato (ATP) a través de la inducción de enzimas glucolíticas que optimizan la utilización de sustratos. Gran parte de este proceso adaptativo es regulado a nivel de la transcripción de genes que se encuentran involucrados en la respuesta a la privación de oxígeno.
Durante la organización de la respuesta celular a la hipoxia son activados factores transcripcionales tales como el complejo activador de proteína-1 (AP-1), factor de crecimiento de respuesta temprana -1 (Egr-1), NF-κB y factores inducidos por hipoxia (HIF). Todos ellos participan en la activación de genes que mantienen la homeostasis vascular12.
El factor de transcripción más estudiado es el HIF-1, heterodímero formado por las subunidades HIF-1α y HIF-1ß, cuya actividad biológica está determinada por la expresión de la primera. Bajo condiciones de normoxia, HIF-1 es hidroxilado por la enzima prolil-hidroxilasa que promueve su unión al complejo Von Hippel-Lindau ubiquitina ligasa (VHL) permitiendo su ubiquitinación y posterior degradación por el proteasoma.
Estudios in vitro muestran que en condiciones de hipoxia, HIF-1 aumenta su estabilidad proteica y potencia transcripcional ya que las enzimas responsables de su metabolización, prolil-hidroxilasa y asparaginil-hidroxilasa, se encuentran inactivas. Algunos autores proponen a estas enzimas como los sensores celulares de la hipoxia13 (fig. 4)
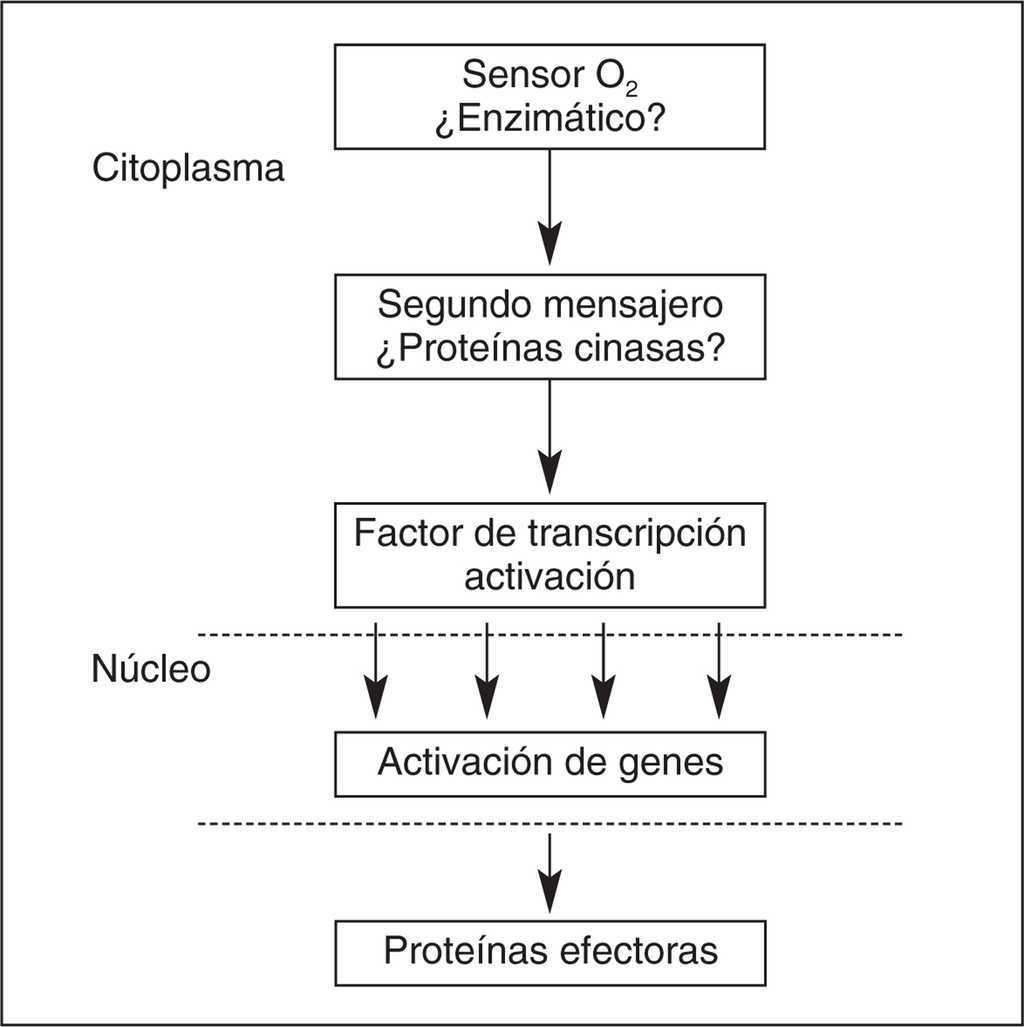
Figura 4. Activación de la transcripción por estímulo hipóxico.
En estas circunstancias HIF-1 se acumula en el núcleo y es responsable de la activación de los genes de eritropoyetina, factor de crecimiento del endotelio vascular (VEGF) y de enzimas glucolíticas y transportadores de glucosa que controlan el ajuste metabólico durante la generación de energía en el ambiente hipóxico. Esta adaptación metabólica involucra una mayor captación de glucosa a través del incremento en la expresión de un transportador de glucosa independiente de insulina (GLUT-1) y la inducción de varias enzimas glucolíticas que mejoran la utilización de sustratos12.
Alteraciones del endotelio ocasionadas por el insulto hipóxico/isquémico
Función de barrera
Muchas de las funciones homeostáticas del endotelio quiescente son mantenidas por señales intracelulares mediadas por nucleótidos cíclicos. Uno de ellos, 3', 5'-adenosin monofosfato cíclico (AMPc) generado por activación de la enzima adenilato ciclasa secundario a la estimulación de receptores de membrana acoplados a proteína G heterotrimérica, participa en el mantenimiento de la arquitectura normal del citoesqueleto de actina permitiendo al endotelio mantener su característica de barrera permeable selectiva (difusión restringida). En condiciones de hipoxia la disminución del contenido intracelular de AMPc ha sido implicada como mecanismo responsable de cambios estructurales que llevan a la retracción de la célula endotelial y aparición de pequeños espacios intercelulares (1-3 µm). Esto conduce a un fallo en la difusión restringida con la consiguiente fuga paracelular de solutos, proteínas y leucocitos. En el desarrollo de este síndrome de fuga capilar también ha sido involucrado el VEGF que además de promover la angiogénesis aumenta la permeabilidad vascular12.
Fluidez circulatoria
La superficie endotelial es fenotípicamente no trombogénica, situación que facilita el flujo sanguíneo. La hipoxia/isquemia al igual que otros agentes proinflamatorios puede desajustar el delicado balance existente entre los mecanismos pro y anticoagulantes. Estos cambios en las propiedades de la coagulación durante la hipoxia surgen a través de diversos mecanismos: a) exposición directa de colágeno o factor tisular desde la membrana subendotelial que activan la vía extrínseca de la cascada de la coagulación; b) supresión de la expresión de trombomodulina, proteína transmembrana restringida al endotelio que acelera la acción de trombina en la activación de la proteína C anticoagulante; c) exocitosis de los cuerpos de Weibel-Palade, organelos cuyas membranas están tapizadas con P-selectina en cuyo interior poseen factor de von Willebrand, el cual promueve la adhesión de plaquetas al endotelio; d) caída brusca de los niveles de óxido nítrico (NO) que contribuye a la acumulación de plaquetas y trombosis; e) inducción de factor tisular por reclutamiento de monocitos en la pared de los vasos; f) supresión del eje fibrinolítico por monocitos, y g) aumento de la expresión del PAI-112.
Regulación del crecimiento celular
La hipoxia/isquemia modula el potencial mitogénico del endotelio al controlar la expresión de factores que participan en la angiogénesis. HIF activados inducen la expresión de VEGF, óxido nítrico sintetasa endotelial (eNOS), factor de crecimiento derivado de plaquetas (PDGF-B) y otros. No está claro aún cuándo la neoangiogénesis comandada por el endotelio activado en respuesta al insulto hipóxico/isquémico tendrá un significado adaptativo o maladaptativo13.
Modulación del tono vascular
El endotelio privado de oxígeno al inicio genera una respuesta vasoconstrictora transitoria seguida de vasorrelajación; sin embargo, si el estado de hipoxia se prolonga, la respuesta vasoconstrictora se perpetúa. La desendotelización experimental de los vasos sanguíneos anula la vasoconstricción inducida por hipoxia, motivo por el cual se cree que esta vasorreactividad está mediada por factores derivados del propio endotelio tales como anión superóxido, endoperóxido, tromboxano A2 y endotelinas. Estas últimas son expresadas en respuesta a la activación transcripcional mediada por HIF-1 y AP-1.
La privación de oxígeno es capaz de activar la célula endotelial en dos etapas, una inicial rápida independiente de activación génica y síntesis de proteínas que incluye retracción endotelial, liberación de factor procoagulante de von Willebrand y expresión de P-selectina y otra lenta que involucra la transcripción de genes y síntesis proteica de moléculas de adhesión y liberación de IL-1 e IL-8.
Por otro lado, el insulto hipóxico crónico estimula la generación de factores mitogénicos con la consecuente proliferación de células musculares lisas vasculares y remodelamiento de la pared del vaso. Si bien la respuesta vasoconstrictora podría considerarse como disfuncional, también es cierto que ésta resulta fisiológica al inicio de la reparación tisular13.
ENDOTELIO Y REGULACION DEL TONO VASOMOTOR
El endotelio se encuentra por una parte en contacto directo con el torrente circulatorio y acoplado estructuralmente con la célula muscular lisa de arterias y arteriolas. Esta característica le permite controlar el tono vasomotor por sí mismo (mecanismos endotelio dependientes) y a través de la interacción que posee con la célula muscular lisa vascular. Por otra parte, existe evidencia de que las células de los propios parénquimas son capaces, en condiciones de hipoxia, de liberar adenosina, lactato, iones hidrógeno, iones potasio y otras sustancias que pueden inducir vasodilatación al activar canales de potasio ATP-dependientes (KATP) que bloquean la entrada de calcio desde el espacio extracelular vía canales dependientes de voltaje (mecanismos endotelio independientes)6,11.
En primer lugar describiremos brevemente los mecanismos que controlan la contracción de la célula muscular lisa vascular para luego comentar cómo participa el endotelio en la regulación del tono vasomotor y su acoplamiento funcional en la unidad reguladora mioendotelial.
Célula muscular lisa vascular
Las células musculares lisas vasculares mantienen el tono del vaso sanguíneo de manera dependiente de la concentración de calcio citosólico, calcio que proviene tanto de los depósitos intracelulares, como de aquel que ingresa a la célula a través de los canales de membrana activados por voltaje. El calcio intracelular disponible forma complejos con calmodulina y activa una enzima quinasa que fosforila la cadena liviana de miosina. Ésta, una vez fosforilada, adquiere actividad ATPasa lo que permite el desplazamiento de las cadenas de miosina sobre actina, evento medular en la contracción de la fibra muscular lisa.
La célula muscular lisa vascular también posee receptores de vasopresina cuyo bloqueo en modelos animales con shock endotóxico reduce la presión arterial y la tolerancia a la hipotensión. Si bien en condiciones normales vasopresina posee un papel menor en el control de la presión arterial, en los estados de shock su concentración plasmática aumenta considerablemente y contribuye a su regulación. Si la hipotensión se mantiene por períodos más prolongados (una hora) los niveles de vasopresina disminuyen producto de una reducción de sus reservas en la neurohipófisis mediada por estimulación barorrefleja intensa y sostenida14.
Endotelio
Los mecanismos de regulación del tono vasomotor dependientes del endotelio son complejos y dependen del balance existente entre la síntesis de moléculas vasoconstrictoras y vasodilatadoras. Entre las moléculas vasoconstrictoras podemos mencionar endotelinas, tromboxano A2 (TxA2) y factor activador derivado de las plaquetas (PAF).
El vasoconstrictor más potente liberado por la célula endotelial en los estados de hipoxia es la endotelina-1 (ET-1). Una vez liberada se une a receptores ubicados sobre las células del músculo liso vascular donde incrementa el influjo de calcio, activa la proteína quinasa C (PKC) y fosforila la miosina, secuencia de eventos que conducen a vasoconstricción12.
Otras sustancias vasoconstrictoras inducidas por hipoxia son PDGF-B y VEGF vinculadas también, como se mencionó en párrafos anteriores, al proceso de angiogénesis. Estas sustancias son expresadas por la célula endotelial sólo en condiciones de hipoxia y principalmente cuando ésta es de carácter prolongado (más de 24 horas).
Las sustancias vasodilatadoras más importantes liberadas por el endotelio incluyen: factor de relajación derivado del endotelio o NO, prostaciclina (PGI2) y factor de hiperpolarización dependiente del endotelio (EDHF)6,12.
El óxido nítrico es una molécula inestable (vida media inferior a 10 segundos) sintetizada por una familia de óxido nítrico sintetasas a partir de L-arginina. En la sepsis la producción de éste se encuentra aumentada en relación con la mayor expresión de la enzima óxido nítrico sintetasa inducible (i-NOS o NOS II) mediada por citocinas inflamatorias. El NO difunde desde el endotelio a las células musculares lisas adyacentes y estimula directamente la enzima guanilatociclasa soluble e induce vasodilatación al reducir el contenido de guanosín monofosfato (GMP) cíclico intracelular. Este último ejerce su efecto vasodilatador al activar una enzima fosfatasa de la cadena liviana de miosina15.
Por otra parte, el NO puede inducir vasodilatación a través de la activación de canales de potasio KATP y dependientes de calcio (KCa) presentes en la membrana plasmática de células musculares lisas. En ambas circunstancias, la movilización de potasio al espacio extracelular implica hiperpolarización de la membrana plasmática y la consiguiente vasodilatación.
La vasodilatación mediada por los canales KCa es de vital interés ya que una de sus funciones es reducir el efecto de los fármacos vasoconstrictores, situación característica en todos los tipos de shock. El NO puede activar estos canales por medio de nitrosilación directa y por activación de la proteinaquinasa dependiente de GMPc14.
La PGI2 ejerce su efecto vasodilatador al incrementar el contenido de AMPc intracelular por activación de la enzima adenilatociclasa a través de un mecanismo dependiente de receptor que conduce a una modificación de la arquitectura del citoesqueleto de actina.
El EDHF ejercería su efecto por un mecanismo independiente de segundos mensajeros al inducir hiperpolarización de la membrana por apertura de canales KATP con inhibición secundaria de la corriente de calcio vía canales dependientes de voltaje. Se sabe además que PGI2 y NO también modifican el potencial de membrana celular con efecto similar al EDHF (activación de canales KATP)6. Por otro lado, estos canales KATP pueden ser activados también por disminución de la concentración intracelular de ATP, aumento de la concentración intracelular de lactato e iones hidrógeno, sustancias neurohumorales como péptido natriurético atrial, péptido relacionado al gen de calcitonina y adenosina14.
Cabe destacar que los canales KATP se encuentran presentes no sólo en las células musculares lisas vasculares sino también en las células endoteliales sugiriendo que éstas participan en la regulación del tono vascular activadas por moléculas como PGI2 y NO, que ejercen su efecto por medio de segundos mensajeros intracelulares y también por acoplamiento eléctrico de ambas membranas celulares6.
Unidad reguladora mioendotelial
Endotelio y músculo liso vascular se encuentran estructuralmente relacionados por medio de uniones amplias (gap junctions). Es posible entonces que ambos tipos celulares se encuentren además funcionalmente acoplados formando una especie de unidad reguladora mioendotelial. Así, estas uniones amplias permitirían el acoplamiento eléctrico de ambas células, de modo tal que aquellos eventos que modifican el potencial de membrana de la célula endotelial serían captados por las células musculares lisas que responderían modificando también el suyo (hiperpolarización → vasodilatación). Como estas gap junctions son más numerosas en arteriolas de distribución, es posible que este acoplamiento endotelio-músculo liso vascular adquiera mayor importancia en lechos vasculares distales responsables de la distribución del flujo sanguíneo a los distintos parénquimas6.
AGRADECIMIENTOS
A nuestra secretaria Srta. Marta Ramírez por su colaboración en la compaginación y diseño de las figuras.

