


En los últimos años han surgido importantes descubrimientos sobre el papel del dióxido de carbono (CO2) a nivel celular y molecular, y sobre los efectos de la hipercapnia. Esta última puede tener efectos beneficiosos en pacientes con patología pulmonar aguda, como la reducción de la inflamación pulmonar y del daño oxidativo alveolar, la regulación de la inmunidad innata, la defensa del huésped y la inhibición de la expresión de citoquinas inflamatorias. Sin embargo, otros estudios sugieren que el CO2 puede tener efectos nocivos en el pulmón, como retraso en la reparación alveolar tras la injuria pulmonar, disminución de las tasas de reabsorción del fluido alveolar e inhibición de la proliferación de células alveolares. Por lo tanto, la hipercapnia tiene efectos tanto beneficiosos como nocivos y es importante determinar el efecto neto en condiciones específicas. El propósito de esta revisión es describir los efectos fisiológicos e inmunomoduladores de la hipercapnia, considerando sus potenciales consecuencias en el paciente con insuficiencia respiratoria aguda.
Important recent insights have emerged regarding the cellular and molecular role of carbon dioxide (CO2) and the effects of hypercapnia. The latter may have beneficial effects in patients with acute lung injury, affording reductions in pulmonary inflammation, lessened oxidative alveolar damage, and the regulation of innate immunity and host defenses by inhibiting the expression of inflammatory cytokines. However, other studies suggest that CO2 can have deleterious effects upon the lung, reducing alveolar wound repair in lung injury, decreasing the rate of reabsorption of alveolar fluid, and inhibiting alveolar cell proliferation. Clearly, hypercapnia has both beneficial and harmful consequences, and it is important to determine the net effect under specific conditions. The purpose of this review is to describe the immunological and physiological effects of carbon dioxide, considering their potential consequences in patients with acute respiratory failure.
En el paciente crítico con insuficiencia respiratoria aguda sometido a ventilación protectora con volúmenes corrientes bajos (Vc)1-3 se permite la elevación de los niveles de dióxido de carbono (CO2) con el fin de prevenir el daño inducido por ventilación mecánica (VILI). En el pasado, al no tener otras opciones para el tratamiento de la acidosis respiratoria, salvo la utilización de un buffer para corregirla, hemos aceptado la hipercapnia permisiva (HP).
Sin embargo, en los últimos años las técnicas extracorpóreas de decapneización por medio de la eliminación extracorpórea de CO2 (Extracorporeal CO2Removal [ECCO2R]) han ido tomado vigencia, con el objetivo de reducir aún más los Vc con el objetivo de prevenir aún más el VILI y evitar la hipercapnia.
Es en este contexto que el CO2 cobra nuevamente importancia al contar con técnicas que nos permiten reducir sus niveles. Sin embargo, ¿debemos prevenir o corregir la hipercapnia en pacientes con insuficiencia respiratoria aguda grave? En los últimos años se han realizado estudios que tratan de dilucidar el efecto del CO2 como agente biológico con efectos a nivel celular y sistémico, con resultados controvertidos.
El propósito del siguiente artículo es revisar los efectos del CO2 y sus efectos a nivel fisiológico y biológico, así como su papel a nivel clínico en el paciente crítico, centrándonos en el síndrome de distrés respiratorio agudo (SDRA) con el objetivo de poder responder a la pregunta planteada.
Efectos fisiológicosEl CO2 produce una serie de acciones a nivel del organismo con diferentes efectos fisiológicos (ver tabla 1e de material suplementario).
RespiratorioA nivel respiratorio el CO2 cumple un papel importante tanto a nivel de oxigenación como de la mecánica pulmonar.
En modelos experimentales, la hipercapnia moderada (FiCO2 5% [PaCO2=50-60mmHg]) mejora la oxigenación arterial, tanto en pulmones sanos como enfermos4-6, al reducir la heterogeneidad ventilación/perfusión (V/Q). Sin embargo, a nivel clínico se ha observado que la hipercapnia en pacientes con SDRA sometidos a ventilación protectora produce hipoxemia por medio del incremento del cortocircuito pulmonar (shunt intrapulmonar), creando zonas de West3 (V/Q<1) resultado de la combinación de aumento del flujo pulmonar e hipoventilación alveolar7.
Con respecto a la mecánica pulmonar, se ha observado que la hipercapnia produce un aumento de la distensibilidad pulmonar a través de la modulación de la interacción actina-miosina a nivel del parénquima pulmonar8, y posiblemente a través del aumento de la producción y mejora de las propiedades del surfactante9.
En cuanto a lo que se refiere a la función diafragmática, existe cierta controversia con respecto al papel de la hipercapnia. En estudios experimentales se observó que la hipercapnia conservó la contractilidad diafragmática y previno su disfunción, probablemente debido a la disminución tanto de la respuesta inflamatoria como de la pérdida de miosina a nivel del diafragma10,11. Sin embargo, a nivel clínico se ha observado que la hipercapnia produce disfunción diafragmática en pacientes en ventilación espontánea por medio de alteraciones en la transmisión de señales eléctricas de las vías aferentes del nervio frénico12. El impacto clínico de la hipercapnia sobre la función diafragmática aún está por ser definido, en especial en el paciente donde se desea el destete y la liberación de la ventilación mecánica.
Los niveles de CO2 parecen tener un papel a nivel de la resistencia de las vías aéreas por medio de la modulación del tono muscular liso. Sin embargo, el CO2 puede aumentar, disminuir o no tener efecto alguno sobre las resistencias pulmonares. Esta variabilidad de respuesta puede ser debida al sitio de acción donde el CO2 ejerce su efecto. Se ha observado que la hipercapnia a nivel local alveolar relaja los bronquios de pequeño tamaño, efecto producido por la modulación de la entrada de Ca2+ en las células musculares lisas bronquiales13. Sin embargo, la hipercapnia a nivel sistémico produce broncoconstricción mediada por estimulación del nervio vago14.
Efectos hemodinámicosA nivel cardiovascular, la acidosis hipercápnica produce un efecto neto estimulador a través de la activación del eje simpático-adrenérgico, con un aumento del gasto cardíaco a través del incremento de la precarga y de la frecuencia cardíaca, disminuyendo a su vez la poscarga. Por otro lado, la hipercapnia produce efectos depresores a nivel cardiovascular, inhibiendo de forma directa la contractilidad miocárdica15 y de las células musculares lisas16. Estos efectos son producidos de forma independiente a los niveles de pH. Sin embargo, son los efectos estimuladores los que prevalecen sobre estos efectos depresores, traduciéndose en un aumento en el transporte de oxígeno.
Otro mecanismo posible del aumento de la oxigenación sería el incremento de la descarga (unloading) de oxígeno a nivel de la circulación (efecto Bohr). Otro mecanismo plausible sería el aumento secundario del hematocrito17.
Si bien los efectos del CO2 a nivel cardiovascular son de aparente beneficio, a nivel pulmonar la hipercapnia produce vasoconstricción capilar y aumenta la presión arterial pulmonar media, lo cual, añadido a los efectos de la ventilación con presión positiva, conduce a un aumento de la poscarga del ventrículo derecho. La hipertensión pulmonar inducida por la hipercapnia puede contribuir a la aparición de cor pulmonale agudo en el paciente con SDRA, en el cual está presente cierto grado de hipertensión pulmonar, y aumentar la mortalidad18,19.
A su vez, la hipertensión pulmonar podría aumentar el estrés en la pared capilar; por lo tanto, en el paciente con SDRA sometido a ventilación mecánica, el empeoramiento de dicho estrés secundario a la hipercapnia, teóricamente, podría exacerbar la lesión pulmonar inducida por sobredistensión mecánica20,21.
Regulación cerebrovascularEl CO2 es un potente regulador del tono cerebrovascular. Por cada mmHg de cambio de la PaCO2 se produce un cambio de 1-2ml/100g/min de flujo sanguíneo cerebral22.
La acidosis hipercápnica produce dilatación de las arteriolas precapilares del cerebro aumentando el flujo sanguíneo cerebral, y es de particular importancia en el paciente con distensibilidad cerebral disminuida, en el cual el aumento del flujo sanguíneo cerebral puede producir hipertensión intracraneal.
El mecanismo probable por el cual el CO2 produciría esta vasodilatación sería la activación de la isoforma neuronal de la sintasa de óxido nítrico (nNOS), incrementado la producción de óxido nítrico, el cual activa los canales de K+-ATP y K+-Ca a través del cGMP, produciendo una disminución del calcio intracelular y vasodilatación secundaria22.
El CO2 es un potente regulador de la ventilación a través de quimiorreceptores localizados en la porción ventral del bulbo raquídeo. Esto es de especial importancia en el paciente crítico con insuficiencia respiratoria aguda (p.ej., SDRA), en donde el trabajo respiratorio incrementa hasta un 30% la producción de CO223, asociado al incremento del espacio muerto alveolar (VDALV)24. Esta hiperventilación compensadora produce un círculo vicioso con incremento en la demanda de trabajo y consumo de oxígeno sobre la musculatura respiratoria, producción de taquipnea, fatiga y claudicación del paciente, que hace necesario el uso de la ventilación mecánica invasiva como medio de soporte, llevando a una serie de efectos deletéreos producto de su propio uso (p.ej., VILI, disfunción diafragmática asociada a la ventilación mecánica), así como efectos derivados producto de la sedación, de la relajación y de la inmovilización prolongada.
Efectos biológicosDaño inducido por ventilación mecánica (VILI)La hipercapnia tiene potenciales efectos beneficiosos que han sido observados en estudios experimentales de lesión pulmonar aguda, como la reducción en los niveles de mediadores inflamatorios o del daño alveolar oxidativo. Sin embargo, diversos estudios sugieren que el CO2 podría ejercer efectos deletéreos sobre el pulmón, independientemente de los niveles de pH (en la tabla 2e del material suplementario se describen los estudios preclínicos en acidosis hipercápnica. En la tabla 3e del material suplementario se resumen los efectos inmunomoduladores de la hipercapnia). A continuación describiremos los efectos del CO2 a nivel pulmonar.
Efectos positivosDiferentes trabajos han demostrado que la hipercapnia reduce el daño por VILI y que sería consecuencia de una disminución del daño producido por sobredistensión mecánica.
La sobredistensión mecánica alveolar produce deformación de la estructura alveolar. El aumento de tensión y/o la rotura del citoesqueleto y la matriz celular activan mecanorreceptores específicos que envían señales al interior de la célula, produciendo liberación de mediadores inflamatorios. Este mecanismo, sumado a la lesión tisular y al aumento de la permeabilidad, puede empeorar el distrés respiratorio ya existente25,26.
El primer estudio que demostró los efectos protectores de la acidosis hipercápnica en un modelo de VILI fue el de Broccard et al.27. En dicho modelo experimental, pulmones aislados de conejos fueron ventilados con presión inspiratoria pico (PIP) baja (15cmH2O) vs. PIP alta (20-25-30cmH2O) y expuestos a hipercapnia o normocapnia. Los autores observaron que la acidosis hipercápnica disminuyó la permeabilidad microvascular, la formación de edema pulmonar y el contenido de proteínas en el lavado broncoalveolar en el modelo con PIP alta.
En estudios posteriores se ha observado que la hipercapnia en diferentes concentraciones de CO2 (FiCO2 4% [PaCO2=45-50mmHg], FiCO2 12% [PaCO2=80-100mmHg]) inhibe los efectos adversos debidos a la sobredistensión mecánica. Estos efectos protectores se producirían a través de los siguientes mecanismos (fig. 1):
- 1)
Mejoría de la oxigenación, de la elastancia pulmonar y de la permeabilidad vascular, con mejoría histológica de las lesiones pulmonares28,29.
- 2)
Prevención de la activación de la vía de las MAP-kinasas, reduciendo así la producción de mediadores proinflamatorios30-32.
- 3)
Reducción significativa de la apoptosis, del estrés oxidativo y de los marcadores de inflamación, al inhibir la activación de las vías MAP-kinasa y SAPK/JNK a nivel de las células epiteliales alveolares26.
- 4)
Disminución de la respuesta inflamatoria y mejoría de la mecánica pulmonar al inhibir la vía canónica de NF-κB, la degradación del IkB-alfa y la translocación del p65 nuclear25.
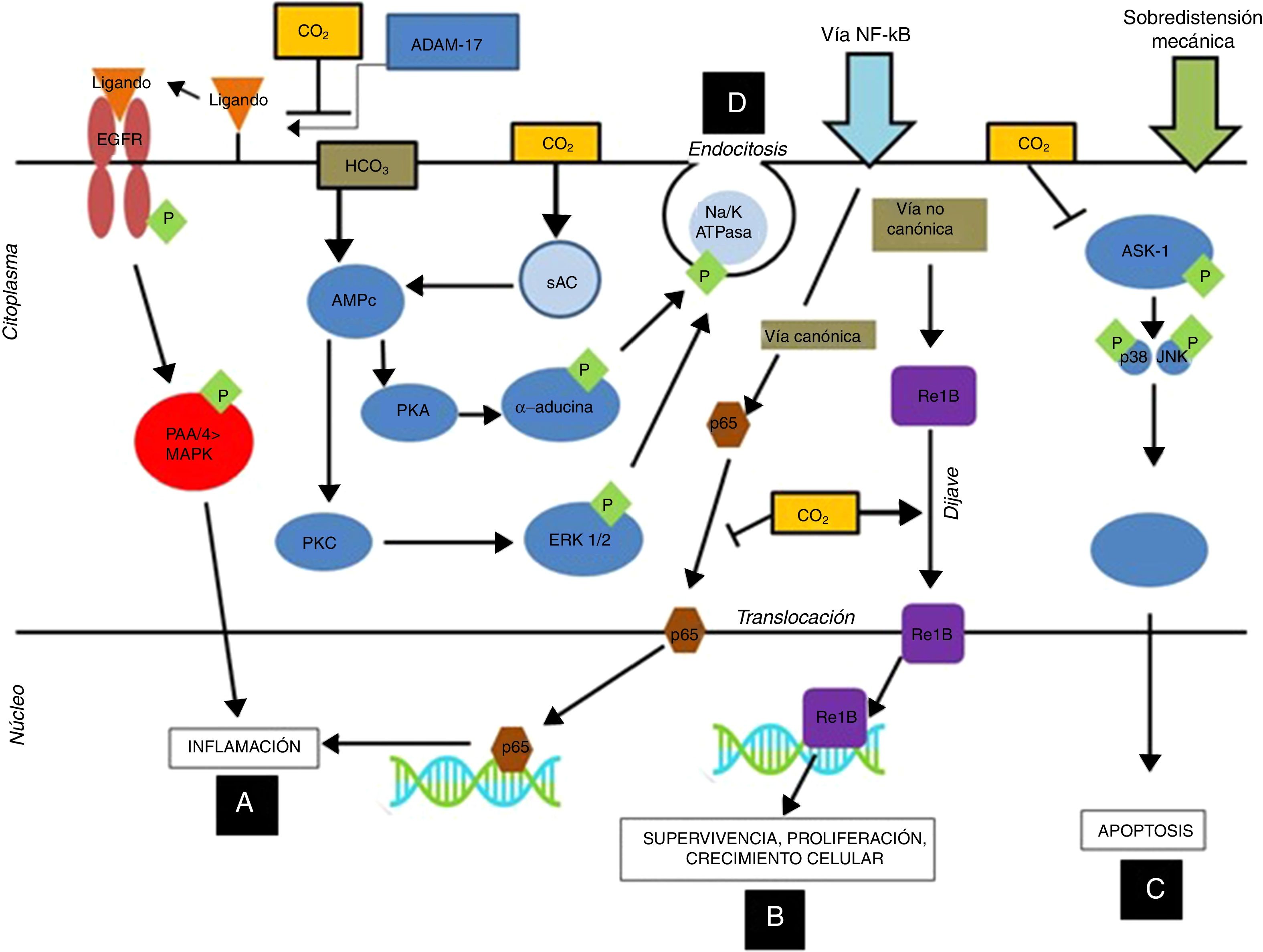
Mecanismos inmunomoduladores del dióxido de carbono a nivel celular: La acidosis respiratoria hipercápnica, por medio de la inhibición del ADAM-17, inhibe la fosforilación de la P44/P42 inducida por sobredistensión pulmonar, disminuyendo así la inflamación nivel de las células epiteliales alveolares (A). Por otro lado, la acidosis hipercápnica estimula la translocación del gen anti-inflamatorio ReIB, y posiblemente disminuye la translocación del p65 al inhibir la vía canónica del NF-kB (B). La acidosis hipercápnica previene la apoptosis producida por sobredistensión mecánica al inhibir la vía MAPK ASK-1-JNK/p38 disminuyendo los niveles de ASK-1, p38, JNK y de la caspasa 3 (C).
La acidosis hipercápnica retarda el aclaramiento del edema a nivel alveolar por medio de la inducción de endocitosis de la bomba de Na-K ATPasa (D).
ADAM-17: ADAM metallopeptidase 17; ASK-1: apoptosis signal-regulating kinase-1; EGFR: epidermal growth factor receptor; ERK: extracellular signal-regulated kinase; MAPK: mitogen-activated protein kinase; NFkB: nuclear factor kappa B; PKA: protein kinase A.
Cortesía de Contreras M. Curr Opin Anesthesiol 2015, 28:26–3769. Copyright © 2015 Wolters Kluwer Health, Inc. All rights reserved.
Al menos un 50% de los pacientes que sobreviven al SDRA experimentan una importante disminución de la capacidad de reserva funcional respiratoria, con limitación funcional y aumento de la morbilidad a largo plazo33,34. Por lo tanto, el proceso de reparación celular posterior al SDRA es de suma importancia en este grupo de pacientes.
La hipercapnia retrasa la reparación del daño epitelial y alveolar tras VILI a través de los siguientes mecanismos (fig. 1):
- 1)
Retraso de la reparación de la membrana alveolar, al disminuir la migración celular dependiente de la vía NF-κB35,36.
- 2)
Disminución del aclaramiento del edema alveolar a través de la inhibición de la bomba de Na+-K+-ATPasa mediante un proceso de endocitosis. Este proceso es independiente del pH y puede ser activado por señales de proteínas del citoesqueleto con receptores para el CO237-40.
El daño por isquemia-reperfusión es el que se produce a nivel tisular cuando el flujo de sangre oxigenada retorna al órgano o tejido tras un período de isquemia, hipoxia o anoxia. Se caracteriza por la activación de una cascada inflamatoria con liberación de citoquinas, neutrófilos, especies reactivas de oxígeno y radicales libres41.
Se produce en diversas patologías del paciente crítico, como el trasplante pulmonar, la embolia pulmonar y el SDRA.
La acidosis hipercápnica ha demostrado que podría atenuar el daño por isquemia-reperfusión a nivel pulmonar:
- 1)
Al preservar la función de la barrera capilar endotelial, disminuyendo su permeabilidad, por medio de la reducción de la actividad de la xantina-oxidasa42.
- 2)
Al atenuar la respuesta inflamatoria, reduciendo los niveles de TNF-α en el BAL y disminuyendo la peroxidación lipídica43-45.
- 3)
Al inhibir la vía de activación NF-κB, disminuyendo la inflamación y la apoptosis a nivel pulmonar46.
La hipercapnia en modelos experimentales de sepsis produce una gran variedad de efectos en el sistema inmune, lo que repercute en el nivel del crecimiento bacteriano.
Los efectos de la hipercapnia en la respuesta inmune se han investigado tanto en estudios in vitro como in vivo:
- 1)
Inhibición selectiva de IL-6 y TNF, citoquinas que juegan un papel clave en la defensa del huésped47.
- 2)
Disminución de la fagocitosis mediada por macrófagos alveolares en modelos animales y en humanos47.
- 3)
Inhibición de la activación de la vía canónica NF-κB, vía que promueve la activación de genes involucrados en la defensa del huésped. Esta inhibición permite la activación de la vía no canónica NF-κB, la cual tiene una acción antiinflamatoria e inmunosupresora48,49.
Se ha observado que la hipercapnia disminuye la capacidad de defensa del huésped tras una agresión de origen microbiano. Esto se ha observado en un modelo murino de neumonía por Pseudomonas aeruginosa sometido a hipercapnia50. En dicho modelo, los ratones expuestos a niveles altos de CO2 presentaron mayor mortalidad e incremento en el número de colonias de esta bacteria a nivel pulmonar y en otros órganos a distancia. Así mismo, se observó una disminución de los niveles de IL-6 y TNF a nivel pulmonar, resultando una menor capacidad de fagocitosis mediada por neutrófilos50.
Hipercapnia y la vía NF-κBLa red del NF-κB está compuesta por 5 familias de monómeros de proteínas (p65/RelA, RelB, cREl, p50, y p52) que forman homodímeros o heterodímeros que se unen al ADN.
Esta red está regulada por dos vías: canónica y no canónica. Estas dos vías controlan los niveles y la activación de los dímeros del NF-κB en respuesta a estímulos, controlando una serie de expresiones genéticas, por medio del reclutamiento de co-activadores o factores de transcripción51.
La hipercapnia parece tener efectos importantes sobre este complejo de proteínas al inhibir la activación de la proteína ReIB por la vía no canónica, que estimula la reparación, la proliferación y el crecimiento celular, e impediría la activación de la proteína p65 (la cual se activa por la vía canónica), la cual ejerce efectos proinflamatorios52 (fig. 1). Por lo tanto, el CO2 ejerce una serie de acciones sobre estas vías a nivel de inflamación y reparación alveolar, así como de inmunidad y defensa del huésped, como se ha detallado en los párrafos anteriores.
Efectos de la hipercapnia en el síndrome de distrés respiratorio agudoHipercapnia permisivaHickling et al.53 fueron los primeros en proponer estrategias de ventilación protectora como rescate en el paciente con SDRA grave con el objetivo de limitar el VILI. Estas estrategias incorporaron las siguientes medidas: 1)disminución de la PIP y la ventilación con Vc bajos; 2)aplicación de PEEP, y 3)aceptar valores altos de PaCO2. Los autores argumentaron en su discusión que «un enfoque alternativo a las estrategias utilizadas de ventilación mecánica sería limitar la PIP reduciendo los Vc, permitiendo la elevación de la PaCO2. Esta se estabilizará a un nuevo nivel superior y la eliminación de CO2 se mantendrá en niveles más bajos de ventilación alveolar, como ocurre con los pacientes con EPOC». Aunque este estudio presenta una serie de limitaciones, la gran diferencia significativa de la mortalidad hospitalaria en favor de las estrategias de ventilación protectora e HP (16% vs 39,6%) dio pie a una serie de estudios prospectivos de ventilación protectora en pacientes con SDRA.
A partir de estos hallazgos, se realizaron 5 estudios clínicos prospectivos aleatorizados para observar el efecto de la ventilación protectora en pacientes con SDRA54-58. Dos de dichos estudios mostraron una reducción significativa de la mortalidad54,57 de la ventilación protectora sobre la ventilación con Vc altos (12ml/kg peso ideal) (ver tabla 4e del material suplementario). Aunque la HP estuvo presente en estos estudios, existen ciertas limitaciones para concluir un efecto protector del CO2, como su alta variabilidad estadística, la no aleatorización de los pacientes a recibir normocapnia vs hipercapnia, además de que el objetivo principal de estos estudios fue demostrar el efecto de la ventilación con volúmenes corrientes bajos (Vc 6ml/kg peso ideal) en la mortalidad en pacientes con SDRA.
Con la intención de analizar si la acidosis hipercápnica ofrecía un efecto adicional a las estrategias de ventilación protectoras con Vc bajos, se realizó un análisis secundario del estudio ARMA59. Se observó que los pacientes hipercápnicos ventilados con Vc de 12ml/kg peso ideal presentaron menor mortalidad que aquellos con niveles normales de CO2 e igual patrón ventilatorio. Sin embargo, en el grupo de pacientes ventilados con 6ml/kg peso ideal no se observaron diferencias de mortalidad en función de los niveles CO2 en plasma. De esta manera resulta difícil sacar conclusiones claras respecto a si la hipercapnia pudiera beneficiar a los pacientes con SDRA más allá de la protección aportada por los volúmenes corrientes bajos.
Recientemente, Nin et al.60, en un análisis secundario de 3 estudios de no intervención prospectivo de cohortes con un total de 1.899 pacientes con SDRA, observaron que los pacientes que desarrollaron hipercapnia definida como PaCO2≥50mmHg dentro las primeras 48h de ventilación mecánica presentaron de forma significativa menor PaO2/FiO2, mayores niveles de presiones meseta, además de un incremento significativo de la mortalidad en UCI (62,5% vs. 49,6%; OR: 1,93; IC95%: 1,32-2,81; p=0,001). Así mismo, la incidencia de barotrauma, disfunción renal y cardiovascular fue mayor en los pacientes con hipercapnia.
Estos hallazgos se encuentran en línea con los reportados por Tiruvoipati et al.61. En este estudio retrospectivo realizado en Nueva Zelanda y Australia, que incluyó más de 250.000 pacientes en un período de 14años, se observó un aumento de la mortalidad de forma significativa en los pacientes que dentro las primeras 24h de ventilación mecánica desarrollaron acidosis hipercápnica (pH<7,35 y PaCO2>45mmHg) (OR: 1,74; IC95%: 1,62-1,88) e hipercapnia compensada(pH 7,35-7,45 y PaCO2>45mmHg) (OR: 1,18; IC95%: 1,10-1,26), comparados con aquellos con normocapnia y pH normal (PaCO2 35-45mmHg y pH 7,35-7,45) (p<0,001).
Aún se necesitan estudios clínicos aleatorizados con un diseño más apropiado para dilucidar el efecto de la HP en el paciente con daño pulmonar agudo.
Espacio muerto alveolarEs importante recordar que los pacientes con SDRA presentan una alteración importante del aclaramiento de CO2 producto del aumento del espacio muerto alveolar (VDALV). El aumento del VDALV en estos pacientes es secundario a alteraciones de la ventilación/perfusión (V/Q), teniendo unidades alveolares ventiladas de forma desproporcionada a la baja perfusión que reciben (V>Q). Esto es resultado de alteraciones de la microcirculación en el flujo sanguíneo, secundarias al daño endotelial, microtrombosis y agregados de detritus celulares62.
El interés por el estudio del espacio muerto en el SDRA fue impulsado por Nuckton et al.24. En un estudio prospectivo de 179 pacientes con SDRA moderado-grave observaron que el aumento del espacio muerto (VD/VT) medido en las primeras 24h del SDRA se asoció de forma independiente a un aumento del riesgo de muerte. Se observó que el VD/VT medio era de 0,54 en los pacientes sobrevivientes vs. VD/VT de 0,63 en los que fallecieron a consecuencia del síndrome. Se registró además un incremento en el riesgo de mortalidad en un 45% por cada aumento en 0,05 del espacio muerto por encima de 0,57. Se observó que la medición del espacio muerto presentó mayor valor pronóstico sobre otras mediciones, como la PaO2/FiO2, distensibilidad pulmonar o severidad de la enfermedad.
Eliminación extracorpórea de dióxido de carbono: futuro prometedorLa razón de tolerar niveles altos de CO2 es permitir Vc bajos, menores niveles de presiones meseta y menor ventilación minuto, con el objetivo de reducir el riesgo de VILI. Sin embargo, hasta el 30% de los pacientes con SDRA presentan evidencia de VILI a pesar de utilizar estrategias de ventilación protectora63.
No obstante, permitir la elevación de CO2 en el paciente crítico con SDRA requiere tomar en cuenta las siguientes consideraciones:
- 1)
Los límites clínicamente aceptables del estudio de Hickling et al.53 (PaCO2 máxima media de 67mmHg, pH medio de 7,20) parecen ser niveles razonables y bien tolerados por el paciente. Sin embargo, mayores niveles de acidosis respiratoria pueden tener efectos no deseados (vasodilatación cerebral, hipertensión pulmonar, arritmias).
- 2)
Si bien se han descrito efectos beneficiosos del CO2 sobre el parénquima pulmonar, la HP no resuelve el problema de regiones no perfundidas del pulmón con VD/VT elevado.
- 3)
La hipercapnia no es el mejor compañero del paciente con SDRA, el cual sufre de una distensibilidad reducida, hipoxia, disnea y alta demanda ventilatoria, en donde se requiere cierto grado de sedación para permitir el comando del ventilador mecánico sobre las necesidades del paciente.
En resumen, la hipercapnia parece ser más una elección de último recurso que un uso terapéutico o rutinario en pacientes con SDRA.
Bajo estos fundamentos, el uso de ECCO2R se ha evaluado como adyuvante de la ventilación protectora, con el objetivo de poder reducir los niveles de Vc a valores inferiores a 6ml/kg peso ideal, denominando esta estrategia «ventilación ultraprotectora» y evitando los potenciales efectos adversos de niveles extremos de acidosis.
En un estudio con 32 pacientes con SDRA de menos de 72h de evolución, Terragni et al.64 observaron una disminución en los niveles de citoquinas inflamatorias en el lavado broncoalveolar en los pacientes sometidos a ventilación pulmonar ultraprotectora (Vc cerca a los 4ml/kg peso ideal) +ECCO2R, demostrando un efecto biológico que evidencia un menor VILI.
En el estudio Xtravent, Bein et al.65 no lograron observar un impacto en la mortalidad de los pacientes con SDRA sometidos a ventilación ultraprotectora +ECCO2R. Sin embargo, en un análisis post hoc del grupo de pacientes con PaO2/FiO2<150 se observó una disminución de los días de ventilación mecánica en los pacientes que recibieron ventilación ultraprotectora (Vc 3ml/kg peso ideal +ECCO2R).
Recientemente, Taccone66 y los miembros del grupo de trabajo de la EuroELSO realizaron una revisión sistemática de la evidencia clínica disponible hasta el momento del uso de ECCO2R en el paciente crítico. Se incluyeron solo los estudios que tuvieran un grupo control. Se encontraron 6 estudios para ser analizados: 3 en EPOC y 3 en SDRA. En estos 6 estudios se incluyeron un total de 279 pacientes, de los que 142 fueron sometidos a ECCO2R con el objetivo de realizar ventilación ultraprotectora. Los dos únicos estudios aleatorizados fueron los del grupo de SDRA. En todos estos estudios se observó una alta heterogeneidad con respecto a los criterios de inclusión, y ninguno de los estudios revisados tuvo el suficiente poder estadístico para concluir un efecto clínico importante, como la estancia en UCI o la mortalidad.
El ensayo SUPERNOVA (NCT 02282657), que ha finalizado su primer reclutamiento piloto de pacientes con SDRA moderado sometidos a ventilación ultraprotectora +ECCO2R, mostrará más datos sobre el uso de ECCO2R en este grupo de pacientes. Así mismo, se encuentra en marcha un estudio clínico aleatorizado con el objetivo de observar la mortalidad a 90días en pacientes con insuficiencia respiratoria aguda hipoxémica sometidos a ventilación ultraprotectora con ECCO2R V-V (NCT 02654327).
Hasta la fecha, la literatura disponible no permite realizar recomendaciones claras sobre el uso de esta técnica en el paciente crítico, siendo su uso por el momento experimental. Por otro lado, las dificultades en la predicción de la progresión del SDRA en una etapa temprana puede limitar el uso de ECCO2R en la práctica clínica.
¿Debemos utilizar un buffer para tratar la acidosis?La utilización de un tampón o buffer para tratar la acidosis hipercápnica sigue siendo una práctica clínica común, aunque controvertida.
La justificación de su uso son los efectos fisiológicos asociados a niveles extremos de acidosis hipercápnica y metabólica (pH<7,10). En particular, la disminución del inotropismo e inestabilidad hemodinámica refractaria a catecolaminas, sus efectos en la función cerebral e inmunológica, y la reducción del metabolismo energético.
Existen dudas respecto al uso de bicarbonato de sodio, buffer utilizado con mayor frecuencia en la práctica clínica. Su administración podría empeorar la acidosis intracelular por medio de la generación de CO2, producto de la reacción del HCO3− con la anhidrasa carbónica, difundiendo de forma pasiva dentro de la célula.
La trometamina (tris-hydroxy-metyl aminomethane [THAM]) se podría considerar una alternativa de elección de buffer en situaciones en las cuales sea necesario tamponar la acidosis hipercápnica. La THAM, al difundir fácilmente en las células, corrige los niveles de pH y reduce los niveles de CO2. Por lo tanto, al corregir los niveles de pH, la THAM podría mitigar los efectos adversos de la acidosis producidos a nivel cardiovascular y recuperar la estabilidad hemodinámica67. Sin embargo, además de las complicaciones asociadas a su uso (irritación, necrosis tisular, hipoglucemia y depresión respiratoria), no resuelve el problema de regiones no perfundidas del pulmón, las cuales producen un aumento del VDALV68.
ConclusionesEl CO2 es mucho más que un producto de desecho del metabolismo celular: es un agente biológico potente que ejerce múltiples acciones a nivel celular con efectos inmunomoduladores a nivel tanto respiratorio como sistémico.
Aunque los estudios preclínicos reflejan un papel beneficioso de la acidosis hipercápnica con respecto a la reducción del daño pulmonar inducido por ventilación mecánica, también existen efectos negativos que podrían reflejar los resultados observados en los estudios clínicos que muestran un aumento de la mortalidad en el paciente con SDRA. Se necesitan más estudios clínicos aleatorizados para determinar el impacto real de la hipercapnia en estos pacientes.
El ECCO2R podría ser una estrategia importante como tratamiento adyuvante en el paciente con SDRA sin hipoxemia grave, permitiendo ventilación ultraprotectora, disminuyendo el riesgo de VILI y controlando los niveles de PaCO2.
Creemos que sería importante identificar niveles de PaCO2 ideales, para equilibrar sus efectos biológicos favorables y desfavorables.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses


