

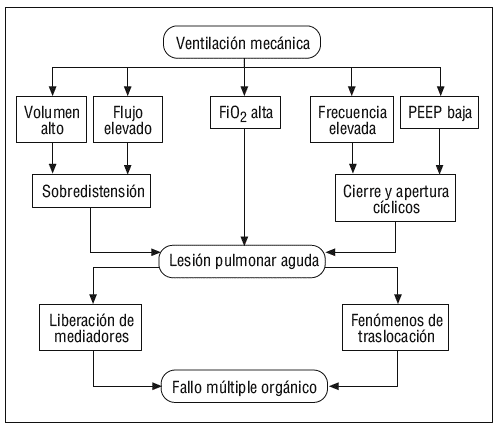


La ventilación mecánica (VM) constituye una importante herramienta en el tratamiento de los pacientes en situación de insuficiencia respiratoria, sin embargo, su aplicación no está exenta de riesgos ni de efectos adversos potencialmente letales. Uno de los cambios conceptuales, quizá el de mayor importancia, que se ha producido en el manejo de los pacientes críticos, es que la propia VM puede dañar al pulmón e inducir o perpetuar la situación de lesión pulmonar aguda (LPA). Este fenómeno se produce tanto en los pulmones previamente sanos, como en aquellos ya previamente dañados, pero es de mucha mayor intensidad, tanto in vivo como en modelos experimentales, en aquellos pulmones con LPA preexistente1.
Existe, además, cada vez mayor evidencia de que la VM no es capaz por sí misma de producir únicamente LPA, sino que surge el concepto de biotrauma, que consiste en que la liberación de mediadores inflamatorios desde el tejido pulmonar dañado colabora en el mantenimiento del síndrome de disfunción múltiple orgánica (SDMO). Este hecho explicaría por qué la mayoría de los pacientes con LPA o síndrome de distrés respiratorio agudo (SDRA) no fallecen en situación de hipoxemia refractaria2, sino en situación de SDMO3,4, y por qué es el fracaso renal agudo asociado al SDRA el principal factor ligado con el riesgo de muerte en este grupo de pacientes5.
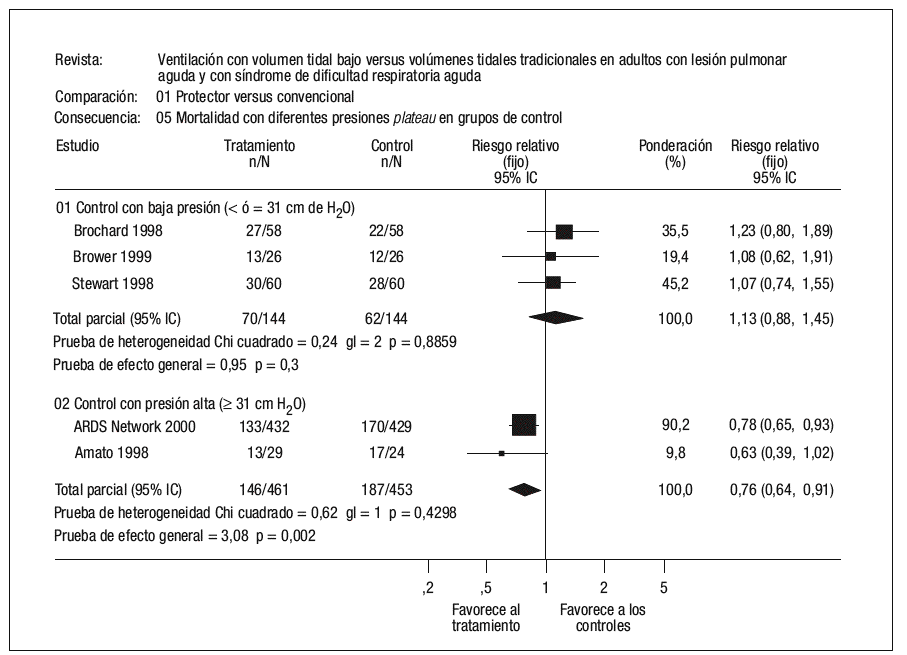
La verdadera importancia de la lesión pulmonar asociada o inducida por el respirador (VILI) ha sido recientemente establecida por los estudios publicados por el Acute Respiratory Distress Syndrome Network6,7, que mostraron una reducción relativa del riesgo de muerte del 22% en aquellos pacientes ventilados con una estrategia ventilatoria protectora del pulmón. La mortalidad atribuible a la VILI sería por tanto de al menos un 9-10%8. Existe, sin embargo, discrepancia en los resultados de los estudios aleatorizados que han valorado el efecto de estrategias ventilatorias protectoras del pulmón en pacientes con SDRA. Una explicación plausible para esta discrepancia es el diferente comportamiento de los grupos control (estrategia ventilatoria convencional) y el diferente nivel de presión en la vía aérea alcanzada en los distintos estudios9. Todos los pacientes que fueron asignados aleatoriamente al denominado brazo protector (bajo volumen corriente) recibieron durante el período de estudio y tras la aleatorización un volumen circulante menor de 8 ml/kg de peso corporal ideal o calculado, y en los cinco estudios la presión media en la vía aérea durante el período de estudio fue inferior a los 30 cm H2O. Sin embargo, el comportamiento de los pacientes que fueron asignados aleatoriamente al brazo control (volumen corriente normal) fue dispar. En los tres estudios en los que no se ha conseguido demostrar una reducción de la mortalidad asociada al empleo de un bajo volumen circulante, se empleó un volumen en el brazo control de 10 ml/kg, mientras en los dos estudios en los que el bajo volumen circulante sí demostró una reducción significativa de la mortalidad, el volumen empleado subió a 12 ml/kg. Esta diferencia en el volumen circulante empleado en los grupos control también se tradujo en una diferencia significativa en la presión plateau de los pacientes durante el estudio, de modo que tanto en el estudio de Amato et al10 como en el estudio de Acute Respiratory Distress Syndrome Network6, la presión fue superior a 35 cm H2O, mientras que en los restantes la presión fue siempre inferior a los 35 cm H2O, e incluso en el estudio de Stewart et al11 esta presión fue inferior a los 30 cm H2O (fig. 1).
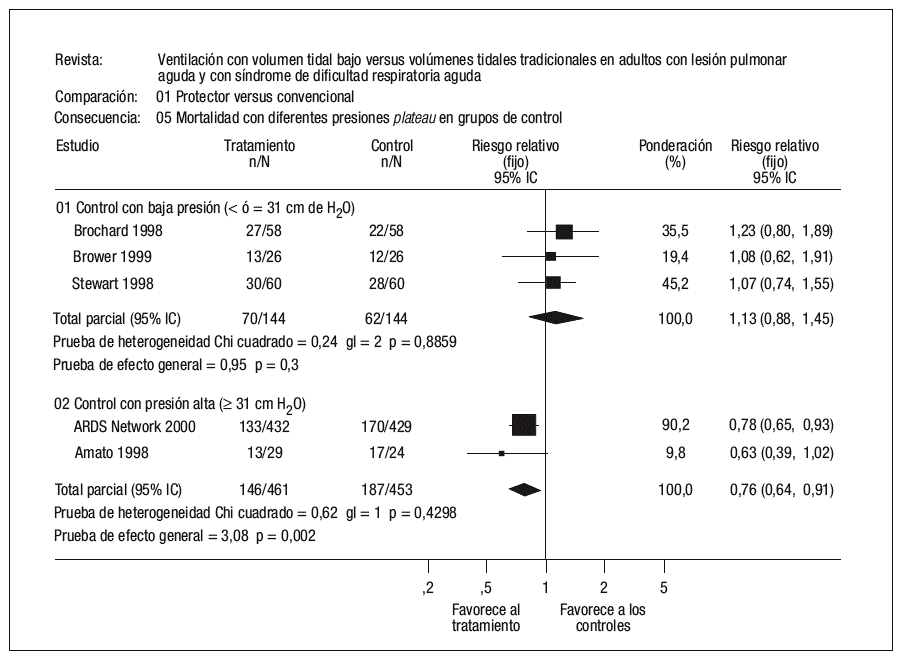
Figura 1. Mortalidad con diferentes presiones plateau en el grupo control. De Petrucci N, Iacovelli W. Ventilation with lower tidal volumes versus traditional tidal volumes in adults for acute lung injury and acute respiratory distress syndrome. The Cochrane Database of Systematic Reviews 2004, Issue 2. Art. No.: CD003844.pub2. DOI: 10.1002/ 14651858.CD003844.pub2.
En su conjunto, los ensayos clínicos concluyen que en pacientes con LPA o SDRA el empleo de un «excesivo» volumen circulante (superior a 10-12 ml/kg de peso) se asocia con un efecto lesional que produce un incremento en el riesgo de muerte de estos pacientes. Sin embargo, cuando se consigue mantener un nivel de presión alveolar inferior a 35 cm H2O, no se ha demostrado un efecto beneficioso por el empleo de un «muy bajo» volumen corriente.
MECANISMOS DE PRODUCCIÓN DE LA LESIÓN PULMONAR INDUCIDA POR EL RESPIRADOR
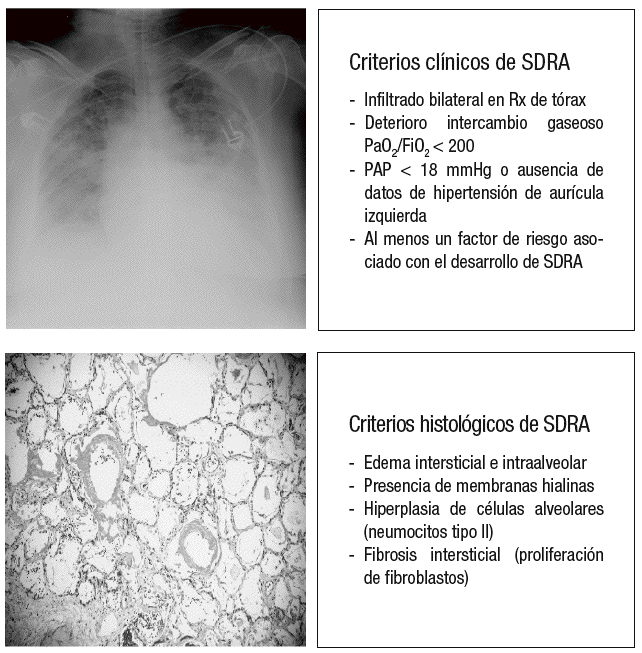
La VILI es iniciado por la aplicación repetida, sobre el tejido pulmonar, de una excesiva tensión y deformación que afecta a estructuras tales como el fibroesqueleto y la microvascularización pulmonar, a las pequeñas vías aéreas distales y a los tejidos yuxtaalveolares, y que es capaz de reproducir no sólo el cuadro clínico de la LPA y el SDRA tal y como fueron definidos por la conferencia de consenso de 199412, sino también la lesión histológica que caracteriza al daño alveolar difuso (DAD), tal y como fue definida en 1976 por Katzenstein et al13, y que constituye el verdadero patrón oro para el diagnóstico del SDRA (fig. 2).
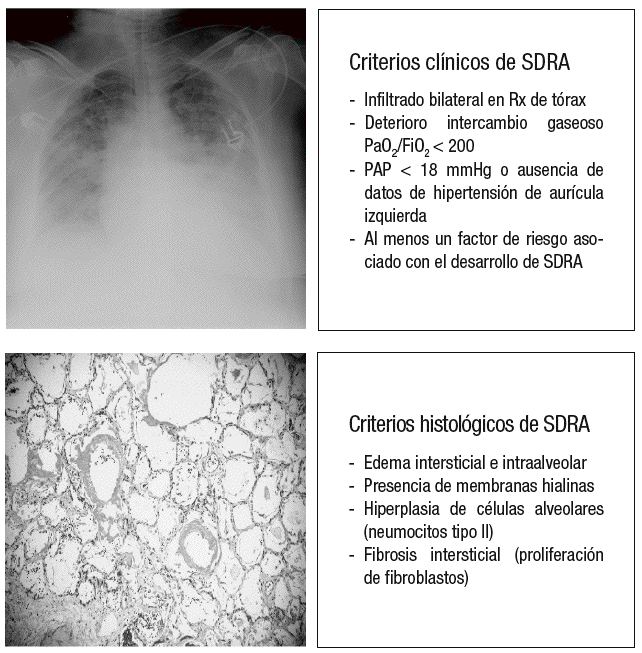
Figura 2. Criterios clínicos e histológicos de síndrome de distrés respiratorio agudo (SDRA). Rx: radiografía; PaO2/FiO2: presión arterial de oxígeno sobre fracción inspirada de oxígeno; PAP: presión arterial pulmonar.
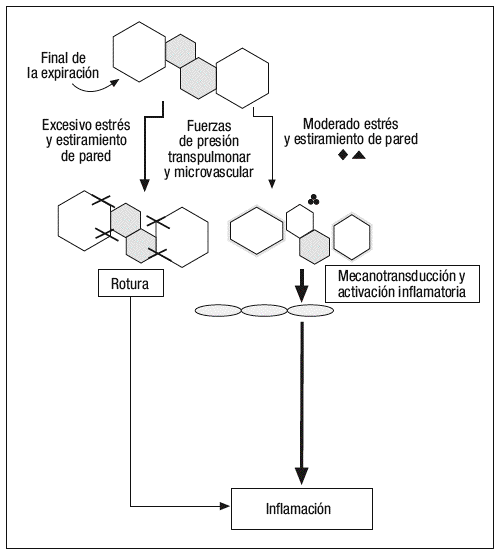
Según un modelo publicado recientemente por Marini y Gattinoni14 (fig. 3), y sin necesidad de que se produzcan presiones intratorácicas elevadas, los cambios en la tensión y deformación de los tejidos pulmonares que se producen en los diferentes ciclos respiratorios son capaces de inducir fenómenos de auténtica ruptura de las paredes alveolares, así como fenómenos de mecanotransducción y liberación de mediadores inflamatorios que pasan a la circulación pulmonar y sistémica, y que son capaces de producir una lesión sobre órganos a distancia del pulmón. No es necesario el empleo de grandes volúmenes corrientes para inducir estos fenómenos de estiramiento pulmonar, debemos tener en cuenta que estas fuerzas de estiramiento se producen una media de 21.600 veces al día (con una frecuencia respiratoria en VM media de 15 respiraciones por minuto) y sobre unos 480 millones de alveolos en unos pulmones de tamaño medio, ya que según mediciones realizadas por Ochs et al15 empleando microscopía electrónica, se ha determinado que en cada milímetro cúbico de tejido pulmonar existen unos 170 alveolos.
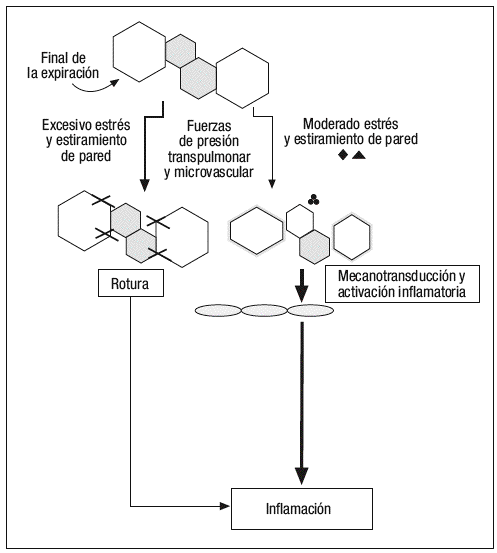
Figura 3. Modelo de producción de lesión pulmonar inducida por el respirador (VILI) propuesto por Marini y Gattinoni.
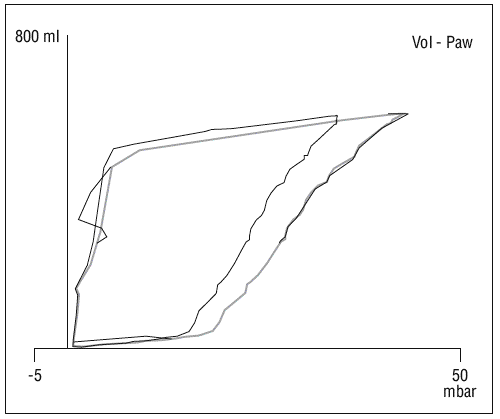
Existe además evidencia de que la disminución de la distensibilidad del sistema respiratorio, descrita en el SDRA, no está relacionada con una disminución de la elasticidad pulmonar, sino que se relaciona primariamente con la pérdida del volumen de gas pulmonar (fig. 4). Así la distensibilidad específica del pulmón es casi normal en pulmones con SDRA y similar a la distensibilidad demostrada en pacientes sanos16,17. También se produce una influencia importante de la distensibilidad de la pared torácica, que está muy disminuida en pacientes con SDRA, de hecho en muchos de estos pacientes existe una elevación de la presión intraabdominal (de cualquier origen) que reduce aún más esta compliance (fig. 5). Estos fenómenos afectan de forma significativa a la presión transpulmonar, que es la fuerza que actúa realmente en la distensión del pulmón18.
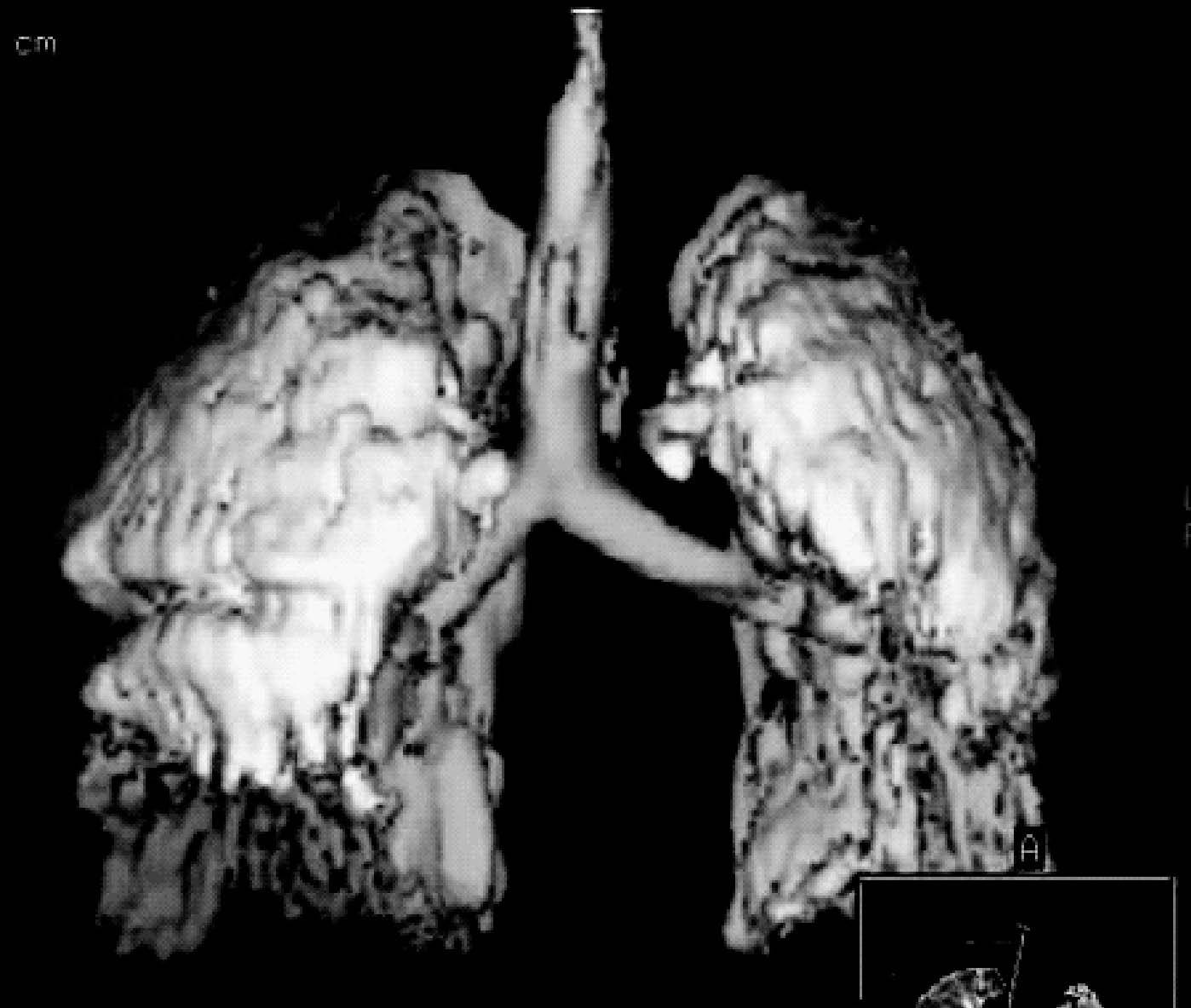
Figura 4. Pérdida de volumen en pulmones con síndrome de distrés respiratorio agudo (SDRA). En la imagen se aprecia una reconstrucción tridimensional de una tomografía axial computarizada (TAC) torácica de un paciente con SDRA y ventilado con una estrategia ventilatoria protectora del pulmón. A pesar del empleo de presión positiva al final de la espiración (PEEP) se puede apreciar cómo el pulmón tiene colapsadas fundamentalmente las zonas correspondientes a las zonas dependientes del mismo (campos posteriores y basales).
Daño pulmonar inducido por el empleo de fracciones inspiradas de oxígeno elevadas
Aunque clásicamente se describe un efecto tóxico sobre el pulmón por el empleo de una fracción inspirada de oxígeno (FiO2) elevada durante períodos prolongados de tiempo, las únicas lesiones que se han podido demostrar son los fenómenos de atelectasia «por reabsorción», con el consiguiente efecto shunt y una bronquitis hiperóxica, sin embargo, en ningún estudio anatomopatológico se ha podido demostrar una lesión característica de DAD en relación con el empleo de FiO2 supuestamente tóxicas. Estas lesiones si han sido objetivadas en estudios realizados en animales de muy pequeño tamaño ventilados con FiO2 de 1 durante períodos de tiempo muy prolongados.
Se piensa que el estrés oxidativo es un mecanismo importante en la producción del daño pulmonar. Este daño se produce mediante la producción de diferentes agentes reactivos con propiedades oxidantes19,20, como el peróxido de hidrógeno, los radicales hidroxilo y los aniones superóxido, que en estos modelos experimentales inducen fenómenos de apoptosis del epitelio alveolar, además de alteraciones de las funciones celulares, fundamentalmente a nivel mitocondrial.
Daño pulmonar inducido por presión excesiva en la vía aérea (barotrauma) y sobredistensión pulmonar (volutrauma)
La relación existente entre la presión en la vía aérea y el daño pulmonar fue demostrada inicialmente por Webb y Tierney21 empleando un modelo de ratas ventiladas durante 1 hora y utilizando diferentes niveles de presión en su vía aérea, con y sin presión positiva al final de la espiración (PEEP). Los animales que fueron ventilados con presiones pico de 14 cm H2O no presentaron ningún cambio histológico en el pulmón, mientras que aquellos ventilados con alta presión (entre 30 y 45 cm H2O) presentaron lesiones consistentes en un importante edema tanto perivascular como alveolar. Exactamente los mismos hallazgos han sido demostrados posteriormente en ensayos con animales de mayor tamaño (conejos y ovejas), aunque en ellos el tiempo de VM preciso para inducir estos cambios histológicos ha sido mayor22-25.
Sin embargo, más que la presión en la vía aérea por sí misma parece que el efecto deletéreo sobre el pulmón está determinado por la sobredistensión del tejido pulmonar, reflejando por tanto más un «efecto de volumen» que un verdadero «efecto de presión». Así, en modelos experimentales de VM en ratas en los que se limita la entrada de volumen circulante mediante el cerclaje del tórax y abdomen, y en los que se induce una sobrepresión importante en la vía aérea (con relativamente bajo volumen), no se ha observado una lesión histológica compatible con LPA26. En cambio, cuando los animales son ventilados en el mismo modelo experimental sin restricción torácica, empleando por tanto un volumen circulante más alto, sí que se produce una lesión pulmonar grave.
Estos resultados se han comprobado también en otras especies27,28. Kolobow et al24, estudiando dos grupos de ovejas sanas anestesiadas y ventiladas, bien con un volumen circulante de 10 ml/kg o bien con un volumen circulante de 50-70 ml/kg, observaron cómo en un período de 48 horas y en las de mayor volumen circulante se desarrollaba un daño pulmonar agudo caracterizado por la alteración progresiva de la mecánica pulmonar y el deterioro del intercambio gaseoso, sin que existiera barotrauma. Este daño se producía con volúmenes circulantes elevados incluso cuando las presiones en la vía aérea eran sólo moderadamente elevadas (30 cm H2O). En la necropsia se encontraron atelectasias pulmonares graves y un aumento del agua extravascular pulmonar, con un incremento significativo del peso pulmonar.
Imai Y et al29 han descrito cómo el empleo de VM, administrada en un modo asistido-controlado, con volúmenes corrientes altos, FiO2 de 1 y con una presión media en la vía aérea de 15 cm H2O, provoca al cabo de cuatro horas un aumento de la cifra de leucocitos polimorfonucleares y de los niveles de factor activador de las plaquetas (PAF), tromboxano B2 (TXB2) y 6 cetoPGF1 en el lavado broncoalveolar (BAL), mientras ocurre una alteración de la mecánica pulmonar, consistente en una disminución de la compliance estática, comparada con los animales con SDRA inducido, ventilados con alta frecuencia.
Daño pulmonar inducido por bajo volumen total (atelectrauma)
Existe evidencia experimental de que lesiones histológicas y cambios inflamatorios compatibles con SDRA pueden inducirse con el empleo en la ventilación de un bajo volumen total, más que por un bajo volumen circulante. Este atelectrauma se produce por el mecanismo de cierre y reapertura cíclico de las unidades alveolares cerradas y da lugar a fenómenos de carácter inflamatorio.
En diferentes estudios experimentales en animales y en humanos30-32 se ha descrito cómo la VM en modelos caracterizados por el empleo de niveles bajos de PEEP da lugar a un incremento de los niveles de marcadores inflamatorios medidos en el BAL. Estos hallazgos consisten en un incremento de los niveles de factor de necrosis tumoral (TNF), interleucina (IL)-8, IL-6, PAF, TXB2 y de la cifra de leucocitos polimorfonucleares que, además, se encuentran activados, como se demuestra por el aumento en la expresión de CD11b (b2-integrina) y la disminución de la expresión de CD62L (L selectina) y de F actina, lo que permite una adhesión leucocitaria inadecuada, aumentando la posibilidad de migración transendotelial.
Colmenero et al33, estudiando el efecto de la PEEP y el empleo de bajos volúmenes circulantes en un modelo de cerdo, también han demostrado cómo el empleo precoz de PEEP previene un aumento de la permeabilidad vascular inducido por la VM.
Tremblay et al34, en un modelo experimental sobre pulmón ex vivo de rata séptica, han descrito cómo la VM con volúmenes circulantes elevados (40 ml/kg) produce un aumento en la concentración de mediadores inflamatorios en el lavado pulmonar. Este efecto está potenciado si el pulmón es ventilado sin PEEP, multiplicando por 50 la concentración de mediadores en el lavado pulmonar. Sin embargo, este estudio puede ser cuestionado porque no tiene en cuenta el posible efecto hemodinámico que puede producir en el propio pulmón y en la incidencia de fenómenos inflamatorios la ventilación con un volumen circulante tan elevado. De hecho, recientemente Broccard et al35 han demostrado en un modelo de pulmón ex vivo de conejo, cómo las alteraciones en la intensidad de la perfusión del pulmón contribuyen a la producción de daño pulmonar inducido por la VM.
Además de la liberación de mediadores inflamatorios a nivel pulmonar, el empleo de estrategias ventilatorias deletéreas en modelos experimentales induce fenómenos de apoptosis celular a nivel pulmonar, e incluso se ha demostrado el fenómeno de traslocación no sólo de IL y mediadores inflamatorios, sino también traslocación bacteriana desde el pulmón36,37.
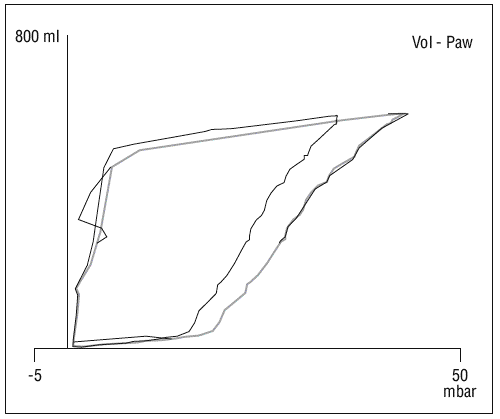
Figura 5. Cambio en la curva P-V en relación con paracentesis evacuadora. En la imagen se aprecian los cambios en la posición de la curva de P-V tras realizar una paracentesis evacuadora en un paciente con ascitis y elevación de la presión intraabdominal por encima de 20 cm H2O.
Daño pulmonar inducido por otros parámetros ventilatorios
Aunque existen pocos estudios, otros parámetros ventilatorios podrían tener influencia en el desarrollo de la VILI, aunque con un efecto mucho menos significativo. Así, el incremento de la frecuencia respiratoria podría dar lugar a un aumento de los fenómenos nocivos sobre el pulmón debido a un mayor estrés de ciclado. En un modelo de pulmón de conejo perfundido ex vivo se ha demostrado mayor edema y hemorragia perivascular cuando éste es ventilado con una frecuencia de 20 rpm que cuando es ventilado sólo con 3 rpm38.
Otro parámetro ventilatorio estudiado es el empleo de un flujo decelerado en lugar de un patrón de flujo constante, asociándose con él una reducción de los fenómenos de sobredistensión pulmonar en las zonas no dependientes, así recientemente Prella et al39 en un diseño experimental en el que miden el grado de aireación pulmonar mediante estudios con tomografía axial computarizada (TAC), demuestran uno de los mecanismos por los que puede ser ventajoso el empleo de la ventilación controlada por presión o con flujo decelerado sobre la ventilación de flujo constante, este mecanismo consiste en una mejor distribución del gas en el pulmón, mediada por un discreto efecto de reclutamiento de zonas pulmonares dañadas, pero sobre todo por evitar fenómenos de sobredistensión en zonas apicales pulmonares, sin que se produzca ningún cambio en la presión alveolar, ni en el intercambio gaseoso (fig. 6).
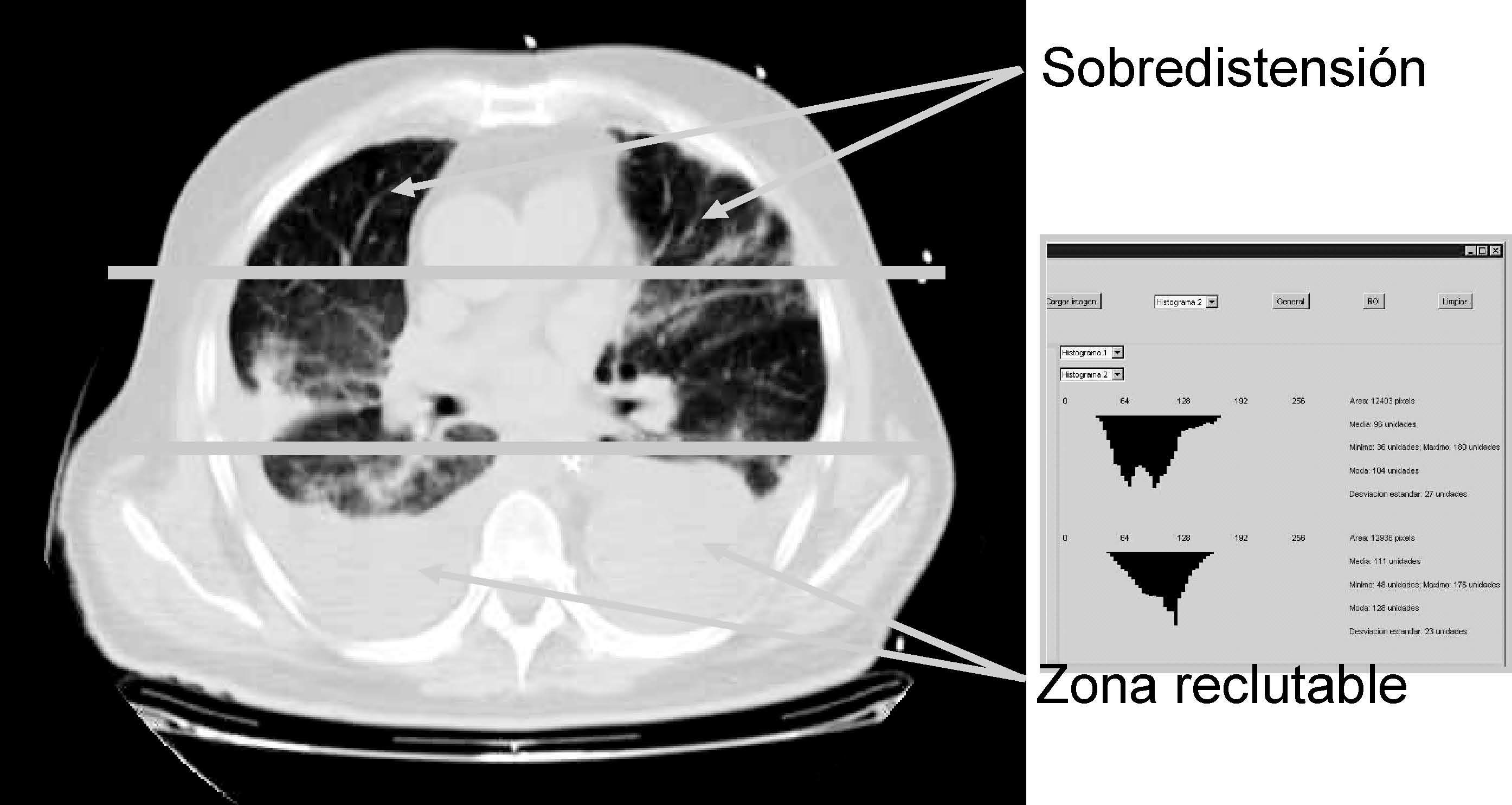
Figura 6. Detección de zonas de compromiso mecánico mediante tomografía axial computarizada (TAC). El empleo de nueva tecnología de radiología y análisis de las imágenes (creación de histogramas de densidades) nos permite analizar zonas del pulmón que podrían participar en la producción de lesión pulmonar inducida por el respirador (VILI).
Además, todos estos fenómenos inductores de lesión pulmonar son mucho más evidentes cuando los modelos deletéreos de VM se aplican en pulmones previamente dañados40 o en modelos experimentales realizados sobre pulmones inmaduros41.
TRANSMISIÓN DE LA LESIÓN PULMONAR A LA CIRCULACIÓN SISTÉMICA
El tejido pulmonar sometido a los mecanismos descritos previamente da lugar a la liberación de diferentes mediadores proinflamatorios, como el TXB2, el PAF y diferentes citoquinas35,40. Esta liberación de mediadores no queda circunscrita al espacio alveolar, sino que, en diferentes investigaciones, se ha demostrado cómo pasan a la circulación sistémica (experimentos realizados tanto en modelos de pulmón de ratones in vivo o ex vivo con perfusión)42,43.
Además, se ha demostrado la traslocación de bacterias o de sus productos desde los espacios aéreos a la circulación sistémica. Estos estudios se han realizado en animales de mayor tamaño (perros) ventilados con ZEEP y con alto volumen tidal36,44, en los que se instilan intratraquealmente poblaciones bacterianas, dando lugar a una bacteriemia.
LESIÓN DE ÓRGANOS A DISTANCIA
A pesar de todos estos conocimientos, de la fuerte sospecha y de los datos indirectos de que la VM aplicada de una manera deletérea podría ser un factor contribuyente en la producción del síndrome de fallo múltiple orgánico, sólo muy recientemente Imai et al45 han demostrado esta asociación.
Los investigadores realizan un modelo experimental sobre conejos de Nueva Zelanda (4-5 kg), sedados, relajados y ventilados mecánicamente a través de una traqueostomía, en los que se les induce un SDRA mediante la administración intratraqueal de ácido clorhídrico. Los animales (n = 24) fueron aleatorizados a dos grupos de VM:
- Grupo 1 (grupo protector): bajo volumen (5-7 ml/kg) y alta PEEP (entre 9 y 12 cm H2O).
- Grupo 2 (grupo deletéreo): alto volumen (15-17 ml/kg) y baja PEEP (0-3 cm H2O).
La frecuencia respiratoria fue similar en ambos grupos, entre 45 y 55 rpm.
El modelo se mantuvo durante 8 horas y posteriormente los animales fueron sacrificados. Ambos modelos de ventilación fueron testados previamente en estudios piloto con el fin de confirmar su efecto sobre el pulmón.
En este estudio se describe cómo los animales del grupo 2 tienen en el aspirado pulmonar y en plasma niveles más altos de proteínas quimiotácticas del monocito (MCP)-1; IL-8 y oncogén regulador del crecimiento (GRO), demostrando una mayor lesión pulmonar en este grupo. Al tiempo, también se encuentra como marcador de lesión de órganos a distancia un aumento significativo del índice apoptótico en las células epiteliales tubulares renales y en las células epiteliales de villis intestinales. Estos cambios también se aprecian en el estudio de estas poblaciones celulares mediante microscopía electrónica.
Resulta llamativo que estos fenómenos no se produzcan en el hígado, y que en el pulmón el índice de apoptosis sea superior en aquellos animales ventilados con una estrategia protectora. Pero es que en ellos, sin embargo, los que son más frecuentes son los fenómenos de necrosis celular (cambios ya descritos en anteriores modelos de isquemia-reperfusión)46.
Estas lesiones descritas son producidas por sustancias presentes en el plasma de estos animales, descartando, por ejemplo, un posible efecto hemodinámico del modelo. Este fenómeno lo demuestran los autores mediante el cultivo de células (LLC-RK1) en plasma de conejos de ambos grupos, al tener de forma significativa mayores fenómenos de apoptosis las células incubadas en plasma de conejos del grupo 2 que las incubadas en plasma de animales del grupo 1.
En el momento actual, y a la luz de los estudios presentados, podemos afirmar que, al menos a nivel experimental, existen conocimientos que indican que los modelos de VM en los que se aplica un alto volumen circulante que produzca fenómenos de sobredistensión de los espacios alveolares, en aquellos en los que se emplean unos niveles bajos de PEEP que permitan fenómenos de colapso alveolar y, por tanto, fenómenos de apertura y cierre cíclicos de unidades alveolares cerradas, y en los que se utiliza una alta frecuencia respiratoria y una alta FiO2, dan lugar a un aumento de la permeabilidad vascular y a fenómenos inflamatorios que conducen a la liberación de mediadores al pulmón y al paso de éstos a la circulación sistémica, siendo capaces de inducir daño a distancia en órganos como el riñón o el intestino (fig. 7). En definitiva, la LPA inducida por el respirador se debe a la producción de un excesivo estiramiento y deformación que afecta a las regiones pulmonares «más sanas» de los pacientes con lesión pulmonar (ya que las regiones colapsadas del pulmón no sufren estos fenómenos de estiramiento y distensión). Esta deformación da lugar a la activación celular, fundamentalmente macrófagos y neutrófilos, a través de la producción de IL-8, las células inflamatorias reclutadas amplifican los fenómenos inflamatorios dentro del tejido pulmonar. Si los fenómenos de estiramiento alcanzan un nivel suficiente se produce ruptura de las paredes alveolares y de los capilares pulmonares y la liberación de los mediadores inflamatorios a la circulación sistémica por mecanismos de traslocación.
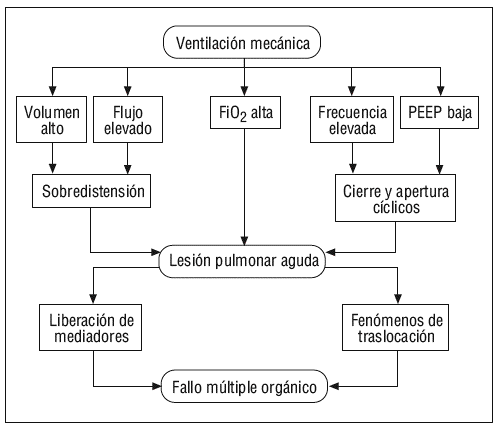
Figura 7. Mecanismo básico del biotrauma. PEEP: presión positiva al final de la espiración.
La mayoría de estos estudios de forma aislada son criticables por ser realizados en modelos ex vivo, generalmente en animales de pequeño tamaño y con modelos de ventilación mecánica exagerados. Por tanto, su aplicación directa sobre lo que verdaderamente ocurre en humanos es difícil. Sin embargo, estos estudios unidos a la evidencia que suponen los estudios llevados a cabo en humanos comparando el efecto de diferentes estrategias de ventilación denominadas «protectoras del pulmón»7,10,11,47-49 hacen un todo que justifica el que se pueda hablar de la VM como contribuyente en la producción del fracaso múltiple orgánico. Sin embargo, hay que reconocer, como citan algunos autores, que los resultados de los estudios no son totalmente consistentes, de modo que no en todos los estudios publicados se observa que la liberación de citoquinas esté en relación con cambios en los parámetros de ventilación, y tampoco es del todo consistente, aún, que esta liberación de citoquinas sea la única productora de la lesión de órganos a distancia50.
¿Es aplicable la teoría de la VILI y biotrauma a pacientes que precisan VM y no tienen SDRA o LPA? Muy recientemente Gajic et al51 presentaron los resultados del análisis de una base de datos generada de forma prospectiva que incluía a 3.261 pacientes en VM, de los que 205 (6,2%) desarrollaron SDRA en más de 48 horas desde el inicio del soporte ventilatorio. Los autores realizaron un análisis multivariable de regresión logística en el que las variables se ajustaron en función de las características del paciente en el momento de su inclusión en el estudio y en función de los factores de riesgo para el desarrollo del SDRA. Encontraron que la aparición de SDRA se relacionaba con el empleo de un volumen circulante superior a 700 ml (odd ratio [OR] 2,6) y con una presión pico srente al 32% en el grupo de pacientes que no desarrolló SDRA. Posiblemente, mientras no se realicen nuevos estudios prospectivos y aleatorizados en pacientes sin LPA que precisen soporte ventilatorio, es desaconsejable el empleo de parámetros ventilatorios que permitan alcanzar presiones alveolares superiores a los 30 cm H2O y volúmenes circulantes superiores a los 10 ml/kg de peso52.
Declaración de conflicto de intereses
Los autores han declarado no tener ningún
conflicto de intereses.

