





INTRODUCCIÓN
La utilización de la ventilación mecánica (VM) como soporte vital lleva varias décadas de aplicación y si bien es imprescindible para salvar vidas, posee numerosas desventajas y complicaciones1.
La lesión pulmonar asociada a la ventilación mecánica (VILI) ha sido durante muchos años sinónimo de barotrauma, entendiéndose como tal la fuga de aire debido a disrupción de la pared alveolar. Sin embargo, los mecanismos de lesión actúan no sólo en el ámbito macroscópico, sino también en el ámbito celular. Los estímulos mecánicos a los que son sometidas las células alveolares desencadenan la liberación de mediadores de la inflamación, proceso denominado mecanotransducción. Así la VM podría iniciar o perpetuar un círculo vicioso de inflamación que conduce a lesiones locales y sistémicas, y desempeñaría un papel importante en el desarrollo de la disfunción multiorgánica. Esta teoría, denominada "hipótesis del biotrauma", cuenta hoy día con gran cantidad de respaldo bibliográfico, tanto en el terreno experimental como clínico2-4.
La interfase alvéolo-capilar es una delgada capa de 0,2-0,3 µm de espesor que se extiende a través de una enorme superficie (hasta 100 m2) sin romperse5. Sin embargo, es llamativo que la mayoría de los investigadores se hayan centrado en los factores que afectan al espacio aéreo durante la ventilación mecánica, como el volumen corriente6,7, la presión pico8,9, la presión meseta10, la presión al fin de espiración (PEEP)11, la frecuencia respiratoria12 y la duración de la VM13, y se hayan desarrollado pocos estudios acerca de los factores que afectan a la microcirculación pulmonar14. Por otra parte, la VM tiene efectos opuestos sobre el diámetro de los vasos alveolares y extraalveolares, generando cambios cíclicos en las resistencias microvasculares, lo que puede contribuir al fallo capilar por aumento del estrés.
Por todo esto en la presente revisión analizaremos el papel de la presión capilar pulmonar, el flujo capilar pulmonar y del endotelio vascular en la generación de la respuesta inflamatoria y en la aparición y progresión de VILI.
PRESIÓN CAPILAR PULMONAR
Definición
Comparando la presión necesaria para impulsar el contenido sanguíneo a través de la circulación sistémica, la misma cantidad de flujo sanguíneo requiere una décima parte de ese gradiente de presión para ser impulsada a través del sistema vascular pulmonar. Alrededor del 60% de la resistencia vascular pulmonar total es precapilar (arterial), mientras que el 40% restante es poscapilar (venosa). Cuando las resistencias vasculares están normalmente distribuidas, la ecuación de Gaar15 permite calcular la presión capilar pulmonar a través de la fórmula: Pcap = PAOP + 0,4 (PAPM-PAOP).
Donde Pcap es la presión capilar, PAOP es la presión de oclusión de la arteria pulmonar y PAPM es la presión media en la arteria pulmonar (fig. 1).

Figura 1. Medición de la presión capilar mediante la técnica de la doble oclusión simultánea. Al detener la circulación las presiones a ambos lados del circuito pulmonar se igualan y determinan la presión capilar. A: imagen de un bloque corazón-pulmones aislado, perfundido y ventilado. Se está realizando la maniobra de doble oclusión simultánea para medir la presión capilar. B: trazado continuo de la presión en la arteria pulmonar y en la aurícula izquierda. Al ocluir simultáneamente ambas ramas del circuito las presiones se igualan y determinan la presión capilar (Pcap).
La precarga ventricular izquierda es habitualmente evaluada a través de la presión de oclusión de la arteria pulmonar, asumiendo que, en ausencia de patología valvular, refleja adecuadamente la presión media de la aurícula izquierda y la presión diastólica final del ventrículo izquierdo. En condiciones fisiológicas, la presión capilar es sólo unos milímetros de mercurio mayor que la presión enclavada, pero las estimaciones de la presión capilar a partir de la ecuación de Gaar pueden carecer de exactitud cuando las resistencias vasculares se alteran16.
Importancia de la presión capilar pulmonar en la formación de edema
La importancia de la exactitud en la determinación de la presión capilar se manifiesta claramente cuando se considera la fisiopatología del edema pulmonar17. El contenido de agua del pulmón normal se aproxima al 80% de su peso húmedo determinado mediante gravimetría y el edema pulmonar es definido por la excesiva acumulación de líquido en el espacio extravascular pulmonar. La clásica secuencia de formación de edema pulmonar sigue el patrón de acumulación inicial de fluido en el espacio intersticial perimicrovascular seguido por la inundación del espacio alveolar, lo cual produce deterioro del intercambio gaseoso por trastorno de la relación V/Q, disminución de la distensibilidad pulmonar y aumento del trabajo respiratorio18.
Fisiológicamente una escasa cantidad de líquidos con proteínas se mueve desde el capilar hacia el intersticio. El sistema linfático drena este fluido hacia el sistema venoso. Cuando la velocidad de filtración de los capilares aumenta, puede exceder la capacidad de remoción máxima de los linfáticos, produciéndose edema intersticial. Cuando el líquido excede la capacidad de almacenamiento del intersticio ingresa a los alvéolos17.
La base fisiológica de la filtración de líquidos y proteínas a través de una membrana semipermeable ha sido propuesta por Starling en 189616: Qf = Kfc · ([Pcap-Pi]-d [COPcap-COPi]).
Donde: Qf: es la velocidad neta de filtración de fluidos, Kfc: el coeficiente de filtrado capilar (producto de conductancia hidráulica de la membrana por el área de superficie de filtrado), Pcap: la presión hidrostática microvascular, Pi: la presión hidrostática intersticial, d: el coeficiente de reflexión de permeabilidad de las proteínas en relación con el agua, COPcap: es la presión coloidosmótica proteica microvascular y COPi: la presión coloidosmótica proteica intersticial.
La ecuación de Starling permite clasificar el edema pulmonar en sus dos variedades principales: edema hidrostático y edema por aumento de la permeabilidad18.
1) El edema pulmonar hidrostático se produce por aumento de la filtración capilar atribuible a cambios en los gradientes de presión hidrostática y osmótica en la circulación pulmonar, cuando las constantes de barrera (Kf y d) no se han modificado.
2) El edema pulmonar por aumento de la permeabilidad de la barrera alvéolo-capilar, como en el síndrome de distrés respiratorio agudo (SDRA), se produce cuando se eleva la tasa de filtración pulmonar asociada a un aumento de la conductividad hidráulica (Kf) o disminución del coeficiente de reflexión osmótica (d) de la pared vascular, a pesar de que los gradientes de presión hidrostática y oncótica no se han modificado.
Los cambios en las constantes de barrera determinan pérdida de la capacidad de la barrera de restringir el movimiento del agua y solutos en la microcirculación pulmonar, así el líquido filtrado suele ser más rico en proteínas en el edema pulmonar de permeabilidad aumentada comparado con la variedad hidrostática pura18.
En condiciones de flujo distinto de cero, el gradiente de presión determina la resistencia, y la distribución longitudinal de los gradientes de presión es directamente proporcional a la distribución longitudinal de las resistencias19. El incremento de las resistencias pulmonares (fundamentalmente poscapilares) que se producen durante la inflamación aumenta la Pcap para el mismo grado de presión en la aurícula izquierda16.
Diversos mediadores de la inflamación, leucotrienos, sustancia P, factor activador de plaquetas y citocinas pueden inducir edema pulmonar puramente hidrostático por aumento de la presión capilar sin producir necesariamente cambios en la permeabilidad capilar. Otros mediadores, como activación de neutrófilos, radicales de oxígeno, factor de necrosis tumoral, etc., producen daño endotelial capilar con el consiguiente aumento de la presión capilar, contribuyendo al desarrollo de edema pulmonar.
Fallo capilar pulmonar por estrés
Mecanismos básicos de la lesión vascular pulmonar
La definición general de estrés es: ESTRÉS = FUERZA/ÁREA; donde el área es el sitio donde actúa la fuerza.
El músculo liso vascular determina la tensión activa de la vasculatura. La función del músculo liso se ve afectada fundamentalmente por la vasoconstricción hipóxica, aunque también por las condiciones hemodinámicas tales como la presión transmural, el tono vasoactivo local y la "fricción "generada por el flujo sanguíneo. Si las condiciones luminales exceden la resistencia elástica de los vasos se generará patología microvascular. A diferencia de lo que ocurre con las fuerzas inducidas por la presión transmural, el estrés generado por los fluidos secundario a fricción (o por la viscosidad del flujo sanguíneo) actúa tangencialmente sobre la pared vascular y el endotelio20. Este tipo de estrés se denomina shear stress.
Los gradientes de presión transmural y la tensión resultante generada en la pared de los vasos está descrita por la ley de Laplace: TENSIÓN = ΔP/r, donde: ΔP: es el gradiente de presión transmural y r: radio.
West et al21 describen las tres fuerzas que actúan sobre los capilares pulmonares:
1) La tensión circunferencial de la pared, causada por el gradiente de presión transmural. Esta tensión es pequeña y comparable con la tensión de la pared alveolar asociada a la retracción elástica del pulmón.
2) La tensión superficial de los alvéolos. Esta fuerza contribuye a mantener los capilares aplanados. La tensión superficial puede ser un factor protectivo frente al fallo capilar por estrés.
3) La tensión longitudinal de la pared alveolar asociada a la insuflación pulmonar. Esta última cobra importancia en la generación del fallo capilar en caso de sobredistensión.
La distensibilidad de los vasos está determinada por el grosor, composición y grado de contracción del músculo liso. Esta tensión se desarrolla en todos los componentes estructurales de la pared de los vasos, incluyendo el tejido conectivo extracelular, y afecta a todas las células que constituyen el citoesqueleto, la cual es transmitida en los sitios de unión célula-célula y célula-matriz.
Por otra parte, si se considera a los vasos como un cilindro rígido, el shear stress está descrito por la ley de Poiseuille´s:ζ = 4Q x η/π x r3, donde: ζ es el shear stress, Q el flujo y η es la constante de viscosidad del fluido.
El shear estress produce deformación celular con incremento de la tensión del citoesqueleto, aunque la dirección de la deformación difiere de la producida por el gradiente de presión transmural. Si bien, in vivo la viscosidad sanguínea tiende a disminuir con la velocidad, modificándose con la distensibilidad de los vasos, el principal determinante del shear stress que sufre el endotelio capilar es el flujo vascular.
Evidencia experimental del fallo capilar por estrés
En 1984, Rippe et al en un estudio en pulmones aislados de perros encontraron que la presión en la aurícula izquierda debía elevarse hasta 55 cmH2O para producir aumento del coeficiente de filtrado, y estos autores atribuyeron el mayor edema a la apertura de "poros"22. West et al, en una serie de experimentos con pulmones de diferentes especies animales, aislados, perfundidos pero no ventilados cíclicamente, sentaron las bases del conocimiento de la fisiopatología del fallo capilar por estrés. Estos investigadores hallaron que un aumento de la presión capilar pulmonar produce alteraciones en la ultraestructura de la barrera alvéolo-capilar, con destrucción tanto del endotelio como del epitelio y paso de eritrocitos al espacio alveolar21,23,24. Si bien las presiones alcanzadas para demostrar este fenómeno de fallo capilar fueron muy elevadas (la presión más baja a la que se observaron cambios estructurales fue de 32,5 cmH2O), es de destacar que todos los experimentos fueron realizados utilizando una presión positiva en la vía aérea constante de 5 cmH2O, sin ventilación mecánica cíclica.
Posteriormente, Guery et al, en un estudio en pulmón aislado, perfundido y ventilado, observaron que la elevación de la presión capilar en presencia de una elevación cíclica de la presión en la vía aérea generaba fallo capilar aun cuando dichas elevaciones eran muy moderadas. Así, la combinación de elevación de la presión capilar con la VM a presiones altas demostró ser peor que una elevación aislada de alguna de dichas variables25.
Influencia de la ventilación mecánica sobre el estrés capilar pulmonar
El sistema circulatorio pulmonar puede considerarse como tres segmentos colocados "en serie": segmento arterial, segmento "intermedio" (capilares) y segmento venoso. El segmento "intermedio", por estar sometido a la influencia de las presiones alveolares, es el que presenta más cambios de resistencia vascular durante la ventilación. Durante la inspiración se produce una distensión del alvéolo, con la consiguiente compresión de los capilares alveolares. Simultáneamente la presión en el intersticio desciende, generando una distensión de los capilares extra-alveolares. De esta manera se genera un aumento del estrés transmural de los capilares extraalveolares y una disminución del estrés transmural de los capilares alveolares. Durante la espiración ocurre lo contrario, aumento del estrés transmural de los capilares alveolares y disminución en los capilares extraalveolares26. Por el contrario, dado que el shear stress que soporta el endotelio de un vaso es inversamente proporcional al cubo del radio del mismo, cuando los vasos se distienden disminuye el shear stress y cuando el radio disminuye (colapso capilar) aumenta (fig. 2).

Figura 2. Microfotografías del pulmón de un conejo que fue sometido a altas presiones transpulmonares. A: fractura capilar con un eritrocito que se está extravasando al alvéolo. B: vista con mayor aumento de la falla capilar por estrés, donde se ven las fibras de colágeno. Por cortesía del Dr. John Hotchkiss.
Fallo capilar pulmonar por estrés en la clínica
Durante la fase inicial del SDRA todas las regiones pulmonares presentan lesión, y la inflamación es difusa y uniforme, pero las alteraciones mecánicas se distribuyen heterogéneamente y cambian con el tiempo27-29. En este primer estadio de la lesión pulmonar existen condiciones de alta permeabilidad y un leve incremento de la presión microvascular pulmonar puede incrementar dramáticamente la formación de edema pulmonar, disminuyendo característicamente la compliancia estática, fundamentalmente por reducción de los alvéolos funcionales30. Distintas regiones alveolares pueden encontrarse ocupadas por edema o estar atelectasiadas, sobre todo en las áreas dependientes, en las que el peso del pulmón suprayacente y la tendencia al colapso de la vía aérea son máximos31. Las consecuencias fisiológicas del edema pulmonar son: compromiso en el intercambio gaseoso, edema en la vía aérea que dificulta el flujo aéreo y la eliminación de secreciones. Por otro lado, la pérdida y alteración funcional del surfactante alveolar favorece la ruptura e incrementa el peso pulmonar, promoviendo la compresión de la pequeña vía aérea, incrementando la tendencia a la apertura y cierre alveolar cíclicos. Aunque los fenómenos de formación del edema pulmonar y de las alteraciones del intercambio gaseoso están relativamente bien descritos, la importancia de la microvasculatura en la generación de VILI es menos entendida14.
Aunque la inflamación posee una importancia fundamental en la pérdida y alteración de la arquitectura pulmonar normal, la elevación de la presión pulmonar transmural puede causar ruptura vascular, como por ejemplo en seres humanos, donde la hemoptisis ha sido descrita en atletas después de haber realizado ejercicios excesivamente pesados32.
FLUJO CAPILAR PULMONAR
El pulmón normal exhibe tres zonas de perfusión, según el modelo tradicional de West33, dependiendo de la relación entre la presión alveolar y la presión arterial y venosa pulmonar. De acuerdo con dicho modelo la presión alveolar es equivalente en todas partes, en condiciones estáticas. En la zona I la presión alveolar es mayor que las presiones arterial y venosa y el flujo de sangre es menor. En la zona II la presión alveolar excede a la presión venosa pero no a la arterial, por lo que el flujo es regular ya que se generan gradientes de presión entre la arteria y el alvéolo. En la zona III la presión arterial y venosa exceden a la presión alveolar, entonces el flujo estaría dominado por gradientes de presión entre arteria y vena y resistencias vasculares. La aplicación de estos conceptos en pulmones con lesión pulmonar aguda no es sencilla, ya que en un mismo microambiente coexisten unidades pulmonares colapsadas, edematosas, inflamadas y fibróticas y, además, las zonas pulmonares cambian a lo largo del ciclo respiratorio y dependen de la programación del ventilador. En consecuencia, a diferencia de lo que ocurre en el pulmón sano donde la barrera alvéolo-capilar está intacta, no queda claro cuál es el umbral de presión hidrostática para la formación de edema en el paciente con lesión pulmonar establecida14.
Broccard et al34,35 han demostrado que la formación de edema hemorrágico durante la VILI ocurre preferentemente en las zonas más dependientes del pulmón, pudiendo ser explicado por varias razones: la disrupción que ocurre durante el estrés aplicado en la ventilación mecánica por la insuflación generada en cada ciclo por el volumen corriente amplía la interfase de cierre-apertura del tejido pulmonar. Una similar línea de razonamiento puede ser aplicada al pulmón con atelectasia y que es expuesto a ventilación con altas presiones, como por ejemplo en SDRA. Otra forma de explicar esta forma desproporcionada de disrupción vascular son las áreas dependientes pulmonares en posición dorsal que reciben la mayoría del flujo sanguíneo y están sujetas a mayor presión hidrostática. Este incremento de la presión vascular intraluminal amplifica las fuerzas de tensión externas en la microvasculatura y provoca cizallamiento y el inicio de la inflamación. Esta interpretación también se apoya en el hecho de que en modelos de VILI y posterior decúbito prono disminuyen las lesiones en las zonas dependientes36.
Los trabajos de Dreyfuss y Saumon mostraron mayor lesión pulmonar producida por ventilación con presión negativa que con presión positiva37. Esto implica que un mayor flujo sanguíneo se correlaciona con una mayor lesión. Además, demostraron que el efecto protector de PEEP durante la ventilación con altas presiones pulmonares se perdía cuando se incrementaba el flujo sanguíneo con dopamina6.
El grupo de Marini et al12,38-40 ha estudiado extensamente las relaciones entre presión capilar, flujo sanguíneo y VILI. De esta manera, Broccard et al38 establecen tres grupos de observación: baja presión en la vía aérea y baja presión en la aurícula izquierda y alta presión en la vía aérea y presión normal en la aurícula izquierda, alta presión en la vía aérea y baja presión en la aurícula izquierda, concluyendo que una reducción en la presión en la aurícula izquierda promueve el incremento de la resistencia vascular probablemente por colapso vascular, el cual se incrementa durante la insuflación pulmonar. Cuando la presión en la arteria pulmonar se incrementa y el pulmón se "desinfla", los vasos alveolares se reexpanden. La excesiva disminución de la presión en la aurícula izquierda puede favorecer la apertura y cierre cíclico de los capilares, generando una falla estructural que explicaría la correlación entre los cambios en la resistencia vascular y la alteración en la permeabilidad vascular con la consecuente formación de edema38.
Hotchkiss et al39 concluyeron que la exposición de pulmones a elevación cíclica de la presión en la arteria pulmonar, en ausencia de ventilación mecánica, genera menos edema y menor hemorragia alveolar y perivascular que aquellos pulmones expuestos a similares presiones en la arteria pulmonar, pero sometidos a ventilación mecánica. Así, los cambios cíclicos en la presión de los vasos perivasculares, extraalveolares, precapilares pueden ser importantes en la génesis de VILI, el cual será atenuado al restringir al máximo el gradiente de presión transpulmonar39.
En otro estudio del mismo grupo establecieron tres grupos de estudio en bloque cardiopulmonar aislado de conejo, combinando presiones en la arteria pulmonar de 20 y 35 mmHg y frecuencia respiratoria de 3 y 20 ciclos por minuto, concluyendo que la disminución de la frecuencia respiratoria puede mejorar los índices de daño pulmonar. Paralelamente, al disminuir el número de veces que se produce el colapso y la apertura cíclicos, la disminución de la presión pico de la arteria pulmonar y la limitación en el flujo sanguíneo disminuyeron el daño pulmonar asociado a la ventilación mecánica12.
Sobre la hipótesis de que la perfusión pulmonar puede alterar la extensión del daño alveolar inducido por la ventilación mecánica, Broccard et al40 realizaron un estudio aleatorizando la velocidad de perfusión de pulmones aislados, y encontraron que flujos de 600 ml/min y 900 ml/min se asociaron a la disminución de la compliancia pulmonar y elevado edema pulmonar (manifestado en la ganancia de peso) y hemorragia (manifestado por histología) comparado con el grupo de flujo sanguíneo bajo (300 ml/min) (fig. 3).
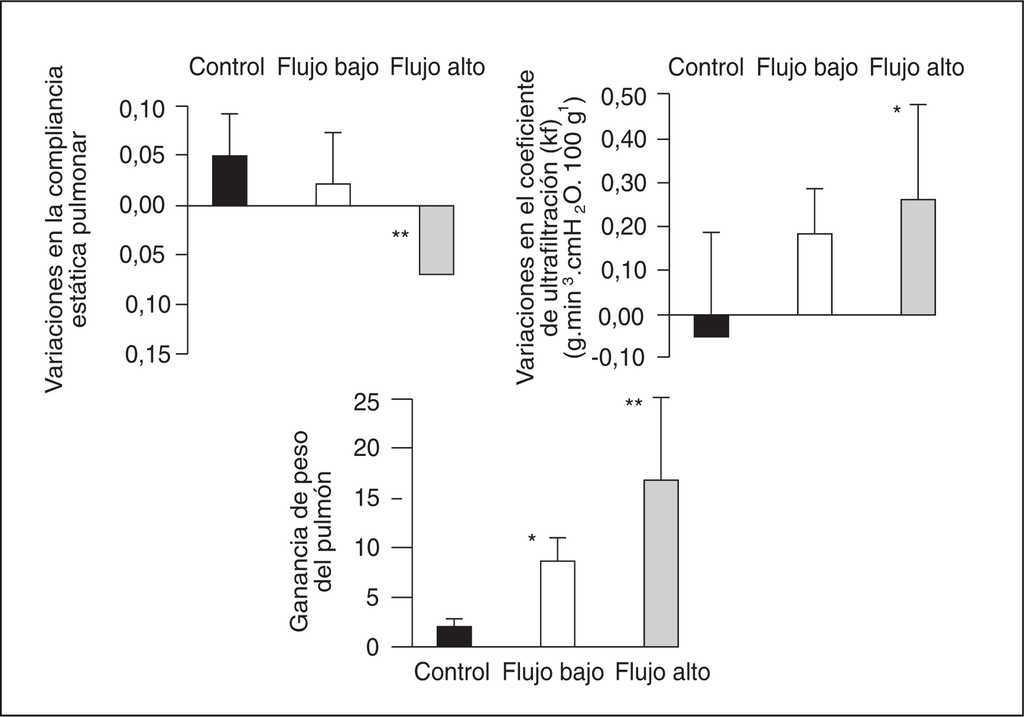
Figura 3. Cambios en la compliancia pulmonar, en el coeficiente de ultrafiltración (Kf) y en el peso del pulmón. Tomada con permiso de la referencia 40. *p < 0,05 para la comparación con el grupo control. **p < 0,05 para la comparación con los grupos control y flujo bajo.
En conjunto, los resultados de estos estudios sugieren que los patrones hemodinámicos pueden interactuar con las presiones de la vía aérea en el desarrollo de VILI sugiriendo que los diferentes patrones de ventilación deben ser considerados, al igual que las diferencias hemodinámicas, por lo que estas conclusiones deberán tenerse en cuenta durante las intervenciones médicas en las que se incrementa el flujo sanguíneo a valores suprafisiológicos en pacientes gravemente enfermos41. Los pacientes con sepsis y SDRA que presentan un volumen minuto cardíaco elevado (espontáneo o inducido) para favorecer la disponibilidad de oxígeno a los tejidos podrían de forma paralela promover la formación de edema pulmonar42.
EL ENDOTELIO VASCULAR COMO GENERADOR DE RESPUESTA INFLAMATORIA
El estrés mecánico y la tensión sobre los tejidos son factores que actúan en la génesis de diversas entidades clínicas43. Las células vasculares están equipadas con numerosos receptores que permiten detectar y responder a las fuerzas mecánicas generadas por la presión y el shear stress44. El citoesqueleto y otros componentes estructurales desempeñan un papel importante en la mecanotransducción, ya que existen conexiones para transmitir y modular la tensión entre las células en sus sitios de adhesión, las integrinas y la matriz extracelular45.
Aunque es bien conocido que el shear stress regula la estructura y la función endotelial controlando la expresión de genes mecanosensitivos y la producción de factores vasoactivos, aún existen mecanismos moleculares que no están aclarados y generan controversias46.
Los mecanismos conocidos de mecanotransducción que participan en la respuesta inflamatoria endotelial al estrés son:
1) Activación de canales iónicos: la rápida respuesta al estrés de las células endoteliales involucra la activación de canales de K+ y Ca2+ 47.
2) Movilización de calcio intracelular: la iniciación de shear stress también lleva a una rápida generación de inositol trifosfato (IP3) y diacilglicerol (DAG) y la subsecuente movilización de calcio intracelular48.
3) Reorganización del citoesqueleto: la aplicación de estrés sobre las integrinas, las cuales están conectadas con el citoesqueleto, produce deformación celular, con incremento secundario de la tensión. Muchas señales moleculares están asociadas con el citoesqueleto, por lo que es posible que la misma tensión desarrollada pueda provocar per se respuestas químicas a los estímulos mecánicos. Las adhesiones focales, las intracelulares, los puntos de contacto entre la membrana plasmática y el citoesqueleto, la activación de receptores y otros elementos estructurales son capaces de provocar señales químicas que participan en estas respuestas49. La reorganización de los componentes del citoesqueleto requiere una despolarización y una repolarización de actina, eventos observados inmediatamente después de la aplicación del shear stress45.
4) Activación de las mitógeno-activadas proteín- cinasas y de las integrina-kinasas: se ha observado la activación del complejo de proteínas de la familia de las mitógeno-activadas kinasas (MAK) como respuesta al shear stress. La activación de esta cascada depende de la proteín kinasa C, pero es independiente del calcio intracelular. También se observó la activación de la tirosín-integrina-kinasa por shear stress50. Ambas cascadas tienen como efector final la activación y translocación nuclear del factor nuclear kappa-beta (NF-*B).
5) Activación del NF-κB el shear stress genera la activación y translocación del NF-κB al núcleo y la activación transcripcional de los genes bajo su control, entre los cuales se hallan: factor de necrosis tumoral α (TNF-α), varias interleucinas, ciclooxigenasa II, óxido nítrico sintetasa inducida, superóxido manganeso sintetasa, factor tisular de la coagulación, péptido natriurético tipo C, ICAM-1, MCP-1, factor de crecimiento de plaquetas B (PDGF-B) y factor transformante beta51,52.
En resumen, el estrés inducido por el flujo vascular pulmonar genera, vía mecanorreceptores de membrana, la activación del complejo de mitógeno-kinasas que inducen la translocación del NF-kB al núcleo y la liberación de mediadores de la inflamación (fig. 4). Esta respuesta inflamatoria puede contribuir al desarrollo y perpetuación de la lesión pulmonar aguda. Esta lesión pulmonar aguda se produce tanto en el endotelio como en el epitelio y provoca la formación de edema intersticial y, en consecuencia, hipoxemia. Cuando la respuesta inflamatoria localizada en el pulmón se desregula resulta en una amplificación excesiva de la cascada inflamatoria y sobreproducción de mediadores humorales que conducen a la descompartimentalización de la inflamación, la afectación de órganos a distancia y la generación del síndrome de disfunción multiorgánica50,53,54. En este sentido, Haitsma et al53 demostraron que la ventilación mecánica lesiva produce pérdida de la compartimentalización de la respuesta inmune en un doble sentido: tanto si la respuesta comienza en el pulmón como si comienza en el abdomen, al aplicar una ventilación lesiva los mediadores inflamatorios aumentan en la circulación. Esta respuesta disminuye al agregar PEEP. Imai et al55 mostraron en un estudio experimental que una estrategia ventilatoria agresiva puede conducir a la apoptosis de células epiteliales renales y del intestino delgado, acompañada de una elevación anormal de marcadores bioquímicos de disfunción de órganos, comparándola con una estrategia ventilatoria protectora. Por otra parte, Rainieri et al56 hallaron que los enfermos con distrés respiratorio agudo ventilados con alto volumen corriente y baja PEEP presentaron altos niveles de citocinas proinflamatorias en el lavado broncoalveolar y en el plasma, a diferencia de los enfermos ventilados con volumen corriente bajo y PEEP alta. Estos hallazgos han sido reforzados recientemente por Stüber et al57. Estos investigadores examinaron el perfil de los mediadores pre y antiinflamatorios presentes en el líquido de lavado alveolar y en plasma de pacientes con lesión pulmonar aguda ventilados con una estrategia protectora (alta PEEP y bajo volumen corriente) cambiando en forma transitoria a una ventilación con baja PEEP y alto volumen corriente. Como conclusión obtuvieron que en los pacientes con lesión pulmonar aguda ventilados con baja PEEP y alto volumen corriente la liberación de citocinas a la circulación ocurría dentro de la primera hora y que podía ser revertido una vez restituida la ventilación protectora. Por otro lado, recientemente se ha demostrado que la lesión cerebral aguda, por mecanismos aún no aclarados, sensibiliza al pulmón y facilita la lesión pulmonar aguda en un modelo experimental58. En resumen, existe evidencia experimental y clínica que indica que el pulmón no sólo es un órgano diana de la respuesta inflamatoria sistémica, sino que también puede actuar como generador de la misma y, en este sentido, el endotelio vascular pulmonar tiene un papel fundamental que debe considerarse a la hora de diseñar estrategias para tratar y prevenir la lesión pulmonar aguda.

Figura 4. El shear stress que se aplica sobre la membrana celular puede generar ruptura transitoria de la misma, pero también activa los mecanorreceptores de membrana. Ambos mecanismos conducen, a través de la cascada de segundos mensajeros intracelulares, a la activación de las MAKK (mitógeno-activadas cinasa-cinasas). La activación de la kinasa específica del inhibidor del factor nuclear kappa-beta (IkB), al fosforilar a dicho inhibidor, permite la liberación del factor nuclear kappa-beta (NF-kB). Éste se transloca al núcleo y se une a sitios específicos del ADN, estimulando la transcripción de los genes que están bajo su control. El ARNm resultante comanda la síntesis proteica correspondiente y los productos finales son liberados al medio o se integran a la membrana celular, según sea su función. Bajo este control del NF-kB se hallan numerosos componentes de la cascada inflamatoria. TNF-α: factor de necrosis tumoral α. IL: interleucina.
CONCLUSIONES
La presión capilar pulmonar no puede ser medida directamente, aunque debe estimarse adecuadamente dado que ésta es uno de los determinantes centrales de la formación de edema pulmonar. En situaciones especiales, como el SDRA y la sepsis, la permeabilidad se encuentra aumentada y la estimación ajustada de la presión capilar cobra mayor relevancia. Aunque la presión de oclusión de la arteria pulmonar no sustituye a la presión capilar, es esta presión la que guía el manejo de fluidos en el SDRA, a pesar de que en este síndrome el gradiente entre la presión capilar y presión de oclusión de la arteria pulmonar se encuentra incrementado. El flujo vascular pulmonar es un determinante de la lesión pulmonar aguda. Independientemente del nivel de presión capilar pulmonar, el flujo pulmonar elevado se ha asociado a mayor lesión pulmonar. Finalmente, el estudio de los mecanismos intrínsecos de la lesión endovascular por estrés nos ha mostrado la relación existente entre el aumento del shear stress y la generación de respuesta inflamatoria y subsecuente lesión pulmonar aguda. Si bien es difícil extrapolar los hallazgos experimentales a la clínica, parece racional pensar que tomar medidas para disminuir el estrés vascular pulmonar disminuirá la lesión pulmonar aguda y, en consecuencia, podría mejorar la evolución de los pacientes con SDRA.
Financiación:
Josefina López-Aguilar, contrato de Investigador Senior FIS 99/3091. Ana Villagrá, contrato de Investigador en formación FIS 01/F015.
Red GIRA G03/063, Marató TV3 y Mútua Sabadellenca Fundació Privada. Fundació Parc Taulí.

