










Research in critical care patients is an ethical obligation. The ethical conflicts of intensive care research arise from patient vulnerability, since during ICU admission these individuals sometimes lose all or part of their decision making capacity and autonomy. We therefore must dedicate effort to ensure that neither treatment (sedation or mechanical ventilation) nor the disease itself can affect the right to individual freedom of the participants in research, improving the conditions under which informed consent must be obtained. Fragility, understood as a decrease in the capacity to tolerate adverse effects derived from research must be taken into account in selecting the participants. Research should be relevant, not possible to carry out in non-critical patients, and a priori should offer potential benefits that outweigh the risks that must be known and assumable, based on principles of responsibility.
La investigación en el enfermo crítico es una obligación ética. Los conflictos éticos de la investigación en medicina intensiva provienen de la vulnerabilidad del enfermo, que en ocasiones ha perdido o visto reducida su capacidad de decidir, perdiendo así su autonomía. Debemos por tanto aunar esfuerzos para que ni los tratamientos, como la sedación o la ventilación mecánica, ni la propia enfermedad cercenen el derecho a la libertad individual de los participantes en la investigación, favoreciendo las condiciones en que hemos de recabar el consentimiento informado. La fragilidad, entendida como el compromiso de la capacidad para tolerar efectos adversos derivados de la investigación, ha de ser tenida en cuenta en la selección de los participantes. Esta ha de ser pertinente, imposible de aplicar en pacientes menos graves y ofrecer a priori unos beneficios potenciales que superen unos riesgos que han de ser conocidos y asumibles en función de una ética de la responsabilidad.
“Preserve health and cure diseases”, this is how Claude Bernard opens his master piece Introduction to the study of experimental medicine,1 this set the starting point of medical research by promoting the application of the experimental method in this field. In his introduction, he says that, without a doubt, experimenting is harder in medicine than in any other science because, although medicine is an applied science, it is different from other disciplines in that the individual is the action subject.
The right clinical practice and, by definition, the ethical clinical practice, only acquires social legitimacy if it has been proven through clinical research. This change of mindset led us to relegate the practice of medicine doctrine based on benefit intention, to mainly base it on presumptions, personal experiences, and subjective criteria.
The decision-making process in critically ill patients is a complex one, consequently the research conducted in this field is complex too. We know that at the ICU, each patient is unique since his physiopathology, response to treatment, and prognosis are conditioned by several determinant factors; we do not deal with certainties as it happens in other sciences like mathematics, but rather we move forward by conducting estimates on individual patients based on researches conducted on groups of similar patients.
Therefore, we should understand clinical research as an ethical obligation for the sake of the scientific advancement for our future generations. But we should also take into consideration here that it includes risks for the subjects of research being mandatory to protect these individuals with special dedication.
The origin of modern bioethics and the atrocities of clinical research with humansThe need for an ethical regulation of research practices in medicine has its origin in historical aspects that we will develop now but is also justified by the scientific advances; the acquisition of new knowledge; and society having a more active and autonomous role in the management of healthcare. In Bernard's work1 it is established that it is immoral to experiment with a person if his participation can be dangerous, even though the result may be beneficial for the rest of the society. This argument that seems undisputable today and has been widely legislated was not the rule of law in the unfortunate researches conducted in the 20th century that gave birth to modern bioethics, based on the patient's autonomy, and respect for his dignity and individual rights. Recently, we have come to know that clinical trials that were being conducted in India have been interrupted2 after reports of individual rights violations, which eventually leads to the need for legislating research in an effort to protect its participants.
History has provided us with several examples of exploitation of human beings in researches being conducted during WWII in Nazi Germany that were described as crimes against humanity in the Nuremberg Trials.3 The Nuremberg Code Decalogue is a set of ethical principles that, for the very first time, states that it is mandatory to obtain the voluntary consent from the individual who is going to participate in any kind of clinical research. Unfortunately, cruelty in experimentation with human beings was not limited to Nazi Germany and it is very likely that other experiments conducted with humans that violated the person's individual rights have never been made public. In Japan, the experiments conducted by Unit 731 during the first half of the 20th century consisted, among others, of studying the progress of lesions of bombarded prisoners. Living human beings were dissected, frozen, and the effects of inoculating toxic agents, toxins, and the exposure to radiation was studied. In the United States, barely a few decades ago, the experiments conducted at Willowbrook State School from 1958 through 1960 were made public and consisted of inoculating mentally handicapped children with the virus of hepatitis in order to study the effectiveness of treatment. During the early decades of the 20th century, experiments were conducted in the Hebrew Orphan Asylum of New York with children where they would be deprived from vitamins in order to study rickets and scurvy. There is no doubt that these events and their repercussion in the public opinion contributed to the development of bioethics. But if one experimentation with human beings was really something else generating a social debate that set a turning point in the development of ethics in research was the study on syphilis conducted in Tuskegee, AL (USA) from 1930 through 1972; in this research the evolution of syphilis was observed without offering the study subjects any of the available treatments in an effort to see how was the natural progression of the disease; during the aforementioned period of time, researchers published over ten papers in scientific journals.4–10 Consequently, back in the year 1974, the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research was created. Its recommendations on clinical research appear in the Belmont report11 published in 1978. Today it is an inescapable reference in clinical research and one foundational documents of modern bioethics. The Belmont report sets the basic principles that guarantee morally appropriate researches (Table 1); if we apply these principles to research with critically ill patients, we could say that the principle of respect to people sees that all patients should be treated like autonomous people, capable of deliberating based on their personal goals, and therefore making consistent decisions; if their autonomy has been compromised, they should be entitled to protection. The autonomy of critically ill patients needs to be protected because these are especially vulnerable patients who do not have absolute control of the situation, are not empowered, or see their own capabilities weakened.12
The benefit principle should be understood as maximizing any potential benefits while reducing as much as possible all possible damages; this is an important trait in critically ill patients, since their vital risk per se puts into question that research should ever be conducted exposing them to risks when no benefit is expected. This makes this population of patients be totally different from other populations of less serious non-critically ill patients who are less exposed to sustaining research-related damage. Therefore, the acceptable risks should be proportional to the expected benefit. The principle of justice is very important when recruiting the participants that will be part of our research, meaning that the benefits, risks, experimentation costs, and their distribution should be assessed here. The scientific need should be based on the largest possible eligibility criteria.
Ethical conflicts in clinical research with critically ill patientsUnlike what happens in other medical–surgical areas, research in intensive medicine poses a challenge due to the difficulties and ethical conflicts it generates. One considerable part of the measures applied in intensive medicine stand on research studies with improvable degrees of evidence; the lack of research with critically ill patients leads to a lack of knowledge that, in turn, makes the patient be vulnerable to severe conditions. The special protection that the critically ill patient needs should not exclude him from research studies that are essential as a value that needs to be preserved. Table 2 shows the main difficulties, all of them leading to ethical conflicts, of research in intensive medicine.
At one time or another during their ICU stay, and due to the treatment measures needed, or the patient's condition, a considerable number of patients will see their capability to make decisions grow thin; in these situations, obtaining the patient's prior written informed consent is an area of conflict we should analyze. If, from an ethical point of view, we know that the risks that the participants of a research study run should be minimal, it seems complicated to apply such principle to the critically ill patient – characterized by his severity and constant proximity to life-threatening situations. Emergency situations are common at the ICU, and the need to act quickly in a climate of uncertainty complicates the studies conducted under such conditions, generating the debate on how research itself should be conducted. Selecting the subjects of research in order to obtain one representative sample becomes complicated in light of the critically ill patient's complex physiopathology, and the multiple or variable factors that may influence the clinical response and, even, the study participation; we have difficulties giving accurate definitions of major syndromes such as sepsis, or distress. As a matter of fact, we saw huge differences in the prognosis of patients in clinical trial that initially had the same inclusion criteria.13
Informed consent and the clinician-researcher dilemma in intensive medicineInformed consent is based on the principle of respect for people and, from this ethical point of view, it is essential when it comes to clinical research.14 There is no doubt that the abuses seen in clinical research during the last century contributed to the development of informed consent which, today, is a prerequisite of clinical research.
The rights of the participants of a single study should be preserved and, based on their free choice of being part of such research, give them the opportunity to understand the potential benefits and risks involved in their participation. Information should be truthful and understandable, and the participants need to comprehend that the primary goal of every research is not obtaining a direct benefit by just being part of a study, but acquire knowledge instead. At this point, it is necessary to distinguish between clinical practice and clinical research. Both share the same uncertainty, but different ethical justifications (Table 3); the goal of clinical practice is to promote benefits through diagnostic and therapeutic initiatives aimed at the patient, while the goal of clinical research is to acquire knowledge in order to improve the management of diseases. The conviction of study patients that they will obtain a direct therapeutic benefit following their participation was already defined decades ago by Appelbaum et al. as what they called therapeutic misconception (TM).15 Although the frequency of occurrence of the TM in critically ill patients has not been studied yet, today it is widely accepted that this circumstance still happens in serious conditions and healthcare areas whenever research and clinical practice are conducted by the same professional. We should make sure that the participants of clinical research studies understand that these studies are aimed at providing benefits to future patients, in order to acquire scientific knowledge, that interventions will be conducted following the protocol, and that the risk/benefit ratio takes place in a climate of greater uncertainty compared to clinical practice.16
With respect to informed consent, both in clinical practice and clinical research, the TM means misinterpreting that consent is one document signed by the patient in order to meet established legal requirements. The fact that informed consent has not been conducted adequately was already confirmed by studies that showed scarce knowledge on the requirements of research studies in participants who signed their informed consent.17 The written registry is nothing, but a minimal part of a much more integrating and communicative process based on the free decision-making process of our patients. Table 4 shows the necessary requirements to obtain informed consent adequately in critically ill patients.18 The critically ill patient's vulnerability rests in his incapacity to protect himself and, in these situations, some structures should be guaranteed in order to minimize damage and exploitation. Precisely, informed consent is a major medical act that requires truthful oral information or, if necessary, through the media, to eventually be included in the clinical history as a signed piece of paper, and in the digital history as part of the evolution of the individual who agrees to participate in this or that research; it is the legal obligation that provides effectiveness to the protection of the participants while research is being conducted. When it comes to the critically ill patient's capabilities to make decisions, we should discuss a few clichés here. Etymologically, the word disease comes from the Latin word infirmitas, meaning “without strength”; making the assumption that critically ill patients cannot make any decisions due to their actual state is a complete misconception that should go into exile. On the other hand, the assessment of this capacity to make decisions is dynamic, meaning that it should be assessed on an ongoing basis, and depending on the different stages of the patient; it is obvious that the presence of delirium, sedation, hemodynamic instability or the condition itself may difficult the process of effective communication with the patient, yet we should not think that just because the patient is in critical condition he is unable to make any decisions during his stay at the ICU. Patients on mechanical ventilation are not incapacitated to make decisions just because their capability to communicate verbally is limited, and now it is more and more common to take care of patients whose level of sedation allows them to adapt to the support treatments required, so we can communicate with them. The actual advances made in the knowledge of sedation and analgesia have enabled us to administer patients the necessary degree of sedation, so that while on mechanical ventilation, they can still understand what they are being told so they can express themselves.19–21 This is why, in these cases, the informed consent document should be handed to the patient while his representative is present, that is, the person who will eventually have to back up the patient's wishes after his approval and consent.22 As a result, obtaining the informed consent from critically ill patients so they can participate in research studies is worth the effort of assessing the patient's capabilities,23 understanding his situation, and looking for adapted ways to obtain the patient's consent in clinical situations like the aforementioned. Today, there is no validated set of recommendations or guidelines to assess the capabilities of the critically ill patient before giving his consent, meaning that there is variability that needs to be minimized. Fig. 1 shows one proposal on how to obtain informed consent for clinical research purposes from critically ill patients (Fig. 1). It is reproachable both to say that critically ill patients are incapacitated, and then obtain the consent of patients who are not in full possession of their faculties, therefore, turning them into subjects vulnerable to exploitation.
Conditions to obtain informed consent adequately in critically ill patients.
| Have enough capabilities to be able to make decisions |
| Receive complete and truthful information on the clinical research |
| Be able to communicate a decision (not necessarily verbal) |
| Voluntariness (no coercion) |
| In informed consents through legally authorized representatives, reassess after the patient is in full possession of his faculties |
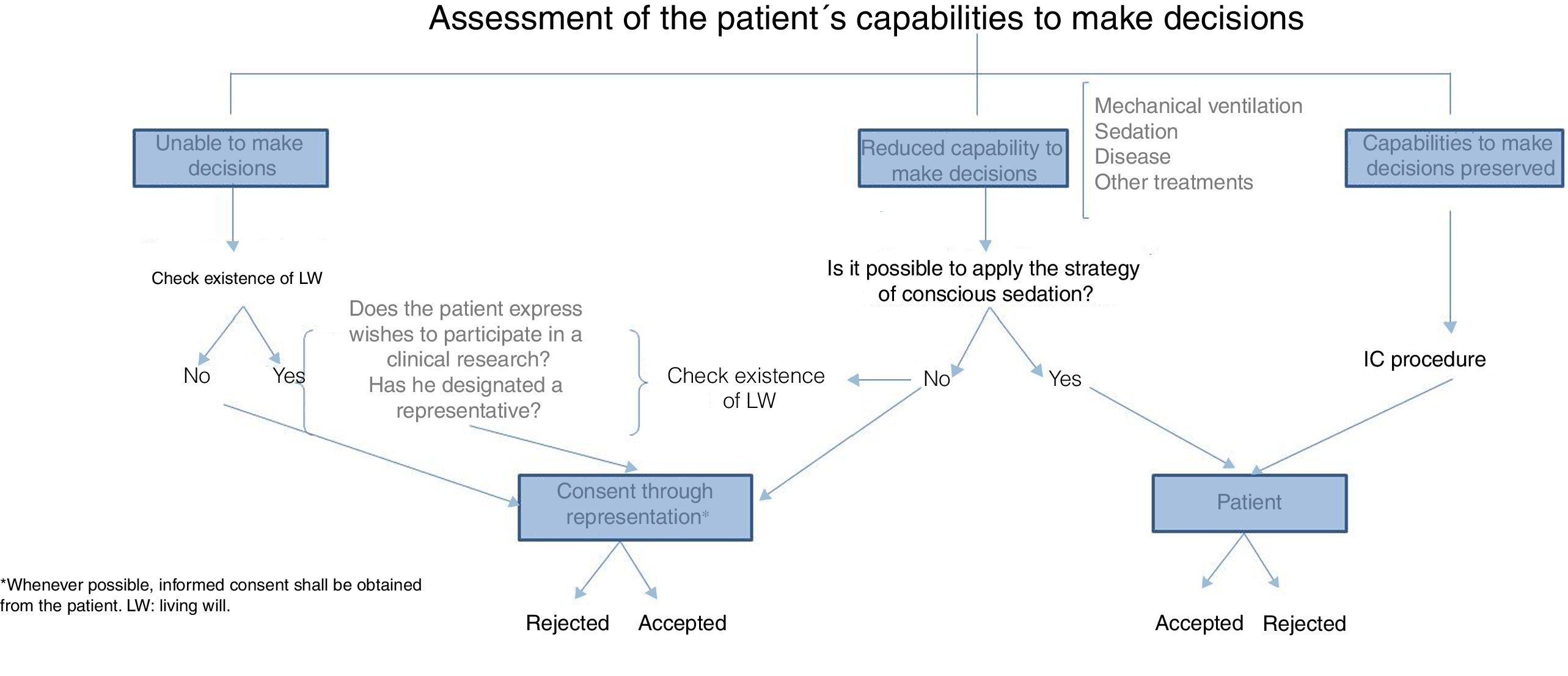
In situations when the patient is unable to give informed consent to participate in a research study, it is widely accepted that, from the ethical point of view, informed consent is obtained through representation, whether through a legal tutor in cases of legally incapacitated patients – uncommon in the ICU setting, or situations of disease-related transient incapacity, or through one representative who is usually someone related to the patient for familial or factual reasons. The search for living wills in patients unable to make decisions at the ICU is mandatory, and the fact that a low percentage of patients have thought about it, does not exclude the need to conduct this search. Once the living will has been made public we have to see not only if the patient's wishes on his prospective participation in research studies are being respected, but also if there is one designated representative.
In consents through representation it is desirable that the representative makes decisions based on what the patient would have decided if he could have made de decision himself; the problem is that these desires are not always known, which is why representations should be conducted in the patient's best interest. The representative needs to be available during the patient's period of incapacity, know what the study is about, and be entitled to withdraw his consent at any time, without prejudice to the healthcare provided to the patient; also, he should not have any conflicts of interest. It is mandatory that, as soon as the patient recovers his capacity to make decisions, he is informed and asked whether he still wishes to be part of the study, or withdraw freely, voluntarily, and without the use of coercion.
The main content that the process of informed consent should include is listed in Table 5. In the case of clinical trials, the content of informed consent is wider than in the rest of studies.
Fundamental content of the informed consent process in clinical research.
| Study primary goal |
| Potential risks derived from clinical research |
| Potential benefits derived from clinical research |
| Diagnostic tests, follow-up and interventions included in the study protocol |
| Identification of researcher/s involved and contact information to be able to contact him/them at any time |
| Possibility to withdraw informed consent at any time during the course of clinical research |
The actual changes society toward a more active role in the decision-making process; the era of new technologies; and the goal of informed consent of establishing a deliberate, communicative process beyond obtaining the participant's signature make it necessary to use simple, clear, easy-to-understand language. Some authors promote the implementation of new types of consent through computing applications, interactive videos, or even through the smartphone, all of them have nothing to do with the classic written document that has a closed structure of long paragraphs with legal and very technical language. The medical literature says nothing about how to implement these models in critically ill patients, but such models stand as an opportunity that should be looked into in the near future, adapting the procedure of informed consent to the reality of the 21st century critically ill patient.24–26
The ethics of healthcare incentives in clinical researchWe could say that no clinical research is designed absolutely non-profit. Incentives not always have to do with something purely financial and they may include fame, prestige, professional promotion, or academic competitiveness. Back in October 19, 2005, UNESCO General Conference promulgated the Universal Declaration on Bioethics and Human Rights27 that in its article 15.2 states: “Benefits should not constitute improper inducements to participate in research”. Researchers can receive money for their participation in research studies for their ideological contribution and research activity. However, it would be unacceptable from an ethical point of view that this was kept from the research. In cases of economic retributions to the study subjects, this cannot be the main motivation of their participation. Transparency in the management of retributions obtained in clinical research needs to be based on an ethics of responsibility that promotes the trust of society in clinical research. Declaring the conflicts of interest should be part of the structure of every research. Today, scientific journals require that the authors of clinical researches provide a declaration of conflicts of interest so that both readers and scientific community are fully aware of such declaration.
Confidentiality and data protectionThe privacy of patients is a fundamental value of people and, as such, it needs to be protected. As a matter of fact, the patient's privacy is protected ever since the beginnings of medicine in the hipoccratic oath that reads: “What I may see or hear in the course of the treatment or even outside of the treatment in regard to the life of men, which on no account one must spread abroad, I will keep to myself holding such things shameful to be spoken about”. In the ancient world, this secret duty should be observed by good professionals, and the term confidentiality became a patient's right back in the 1970s. There are many pieces of legislation on this issue, integrated in research protocols and requested by the RECs for the protection of the study subjects. Personal data should receive confidential treatment, and the same guarantees should apply to biological samples; also, third parties should be allowed to have access to the patient's personal data only if they have the patient's authorization. The boom of the digital era and its increasing application among healthcare institutions and research studies makes it essential to take security systems to the extreme in order to guarantee the confidentiality of the research subjects.
Research ethics committeeIn Spain, there is more than a hundred research ethics committees (REC), and the transfer of healthcare competences to the Spanish autonomous communities translates into different REC accrediting systems, asymmetries in training, and lack of coordination among them.
There have been situations where one same project in a healthcare area with the exact same characteristics and similar research profile has obtained different judgment from the REC involved depending on this or that REC.
RECs conduct ethical and legal methodological assessments of research projects. Also, they play a fundamental role in the follow-up of research, and they can introduce modifications to the project or even interrupt it based on the events that may unfold during research28; this project follow-up role of RECs is not easy to put into practice today. There is evidence of interventions by the committees on clinical trials conducted in critically ill patients. In the United States, in the year 2000, one anonymous letter29 addressed to the U.S. Office for Human Research Protections (OHRP), an administration that regulates local RECs, criticized the design of one prestigious study conducted in critically ill patients with distress,30 mentioning reasons of absence of informed consent to be able to conduct the research. Two years later, the OHRP suspended another study designed by the same taskforce, the ADRS network, on strategies of fluid administration through central venous catheters, and pulmonary artery catheters in patients with distress, claiming that the study groups had not been compared using the best routine clinical practice; also, the OHRP questioned the ventilation procedures of patients,31 leading to a scientific debate with letters and opinions from clinicians, researchers, and bioethics experts on the study design and proving how difficult it is to define standard clinical practice in critically ill patients. Research ethics committees also contribute to the ethical quality of the publications, and today their approval is an indispensable prerequisite for papers to be accepted by any prestigious scientific journals.
Legislation in clinical researchThe Nuremberg Code, designed back in 1947, is considered the very first document to ever regulate clinical research.
Then, another fundamental document for clinical research that is over 50 years old now was published by the World Medical Association (WMA) in its 18th assembly held back in 1964 and ever since it has been reviewed on an ongoing basis. Its last and 7th iteration took place in the General Assembly held in October 2013 in Fortaleza, Brazil. Ever since 2016, the Declaration of Helsinki is complemented by the Declaration of Taipei on the ethical considerations of healthcare databases and biobanks. Back in 1978, the Belmont report was published, whose basic principles were already discussed a few pages ago. The Oviedo Convention became effective in January 2000, and it has been incorporated to the national legal framework as a law. In its 5th chapter, it establishes the requirements needed to be able to conduct studies with human beings and states that there should not be an alternate method to conduct studies of similar effectiveness, that risks should not be disproportionate to the benefits obtained, that research projects should be approved by the corresponding authority and that informed consent should be obtained after providing all the necessary written information. When it comes to critically ill patients, although the document does not address this population specifically but rather disabled people, additional guarantees are established on the issue of expected benefits being a direct benefit for the participants of clinical research; that results from research should not be obtained in people able to consent; that the participant-to-be should not have expressed his rejection to participate in the study; and that all requirements for consent by substitution should be observed.
When it comes to the legal framework of clinical research in Spain, the main pieces of legislation that regulate clinical research are the Organic Law 15/1999, of December 13th, on the protection of personal data32 and Act 41/2002, of November 14th, the regulatory base of the patient's autonomy, rights, and obligations on information and clinical documentation,33 both complementing Act 14/2007, of July 3rd, on biomedical research.28 Although the legal framework on clinical research today is simply huge, there is no regulation that specifically addresses a well-defined population such as critically ill patients; still, the Act of Biomedical Research dedicates Title II (articles 13 through 27) to clinical research involving invasive procedures in human beings.
Scientific fraudThe honorability of clinical researchers should not be jeopardized by the despicable practices of scientific fraud of which a few cases have been disclosed. It is likely that although this is a much more common situation than we think, it is still undisclosed or uncovered. From the ethical point of view, the scientific fraud discredits clinical research in the eyes of society, leading to a climate of disbelief; also, it jeopardizes patients, since the disclosure of results can be erroneously applied to the clinical practice. Fictitious fabrications, falsification of results, and plagiarism are examples of this type of frauds. The true meaning of clinical research is to generate knowledge, and expose the results obtained, even when such results may be unwanted or unexpected by the research team or promoters. Failure to comply is called publication bias,34,35 and, from the ethical point of view, it is unacceptable not only because it takes away valuable information from the scientific community, but also because it exposes, in vain, the participants of clinical research to risks.
It is problematic to replace the true goal of research for a secondary goal, reducing the primary goal to publishing in a scientific journal for the sake of improving the author's own résumé, or receiving incentives from the pharmaceutical industry. The cases of scientific fraud involving Dr. Boldt and Dr. Fujii have been the biggest scandals ever in intensive medicine; also, when one case of fraud is detected, it is not usually an isolated case and the rest of papers published by this author are investigated too. The case of Dr. Joachim Boldt led to withdrawing 88 papers from international journals back in the year 2011.36 Dr. Yoshitaka Fujii had worked for several universities before and had been the author of numerous research papers. The lack of approval from the ethics and data management committes,37 was investigated, and the Japanese Society of Anesthesia recommended the withdrawal of 189 papers on anesthesia and critical care. Fighting scientific fraud is everyone's responsibility: researchers, institutions, scientific journals, research ethics committees, and readers of scientific literature should all take a step forward when something looks suspicious so that it is properly investigated. The ethics committees on publications establishes the guidelines required to promote good practices, from the ethical point of view, in scientific publications,38 and details penalties that go from clarification letters from the authors responsible of minor cases, to the withdrawal of scientific papers by informing other publishers and authorities of the scientific fraud perpetrated in the most serious cases of fraud.
Ethical aspects in the selection of critically ill patients for research purposesUnlike what happens in other groups of vulnerable patients, in Spain there is no specific legislation on clinical research with critically ill patients. The protection of research subjects is, in the first place, the responsibility of researchers and promoters, but it needs to be regulated by an independent group such as the research ethics committees, that will be looking into the quality with which informed consent is obtained, and evaluate the adequate study design, paying special attention to the vulnerability of the critically ill patient in the analysis of the risk/benefit ratio. Table 6 lists the ethical conditions that we should bear in mind in the selection of critically ill patients for research purposes.
Ethical conditions required for clinical research in critically ill patients.
| Impossibility to obtain similar results in clinical research conducted with non-critically ill patients |
| Possibility to obtain benefits following participation in research studies |
| The risks and burdens cannot be disproportionate to the potential benefits |
| Informed consent through the patient or his representative |
| In randomized trials, the patient should be informed on the possibility of selection in different treatment arms of the study |
| In comparative studies, the control group should receive the best standard clinical practice backed by scientific evidence |
From the ethical point of view, clinical research with critically ill patients is a challenge. It is mandatory to protect the generous act that research subjects provide society with by participating in these very necessary research projects; it is the sacrifice of a few for the good of many. On the one hand, we should understand clinical research as an ethical obligation toward future generations and, on the other hand, we should be protecting this type of vulnerable and fragile patients. Ethics aims at what's optimal, while trying to balance the values that come into conflict. The high level of care dependency and state of the critically ill patient compromises his capacity to tolerate adverse events derived from research, which is why studies should be worthy, that is, relevant, and all the potential benefits of clinical research should overcome all of the potential tolerable risks. The obstacles that severe diseases and their treatment pose for the critically ill patient's decision-making process should be fought while being especially respectful to the patient's autonomy. The advances made in the ethics of clinical research during the last few decades have improved the requirements of research itself, but there is still much to do, and we still need to improve the process of informed consent and conduct independent assessments and follow-ups of projects with subjects external to research. Healthcare institutions, research ethics committees, scientific societies, promoters, and researchers should all work together for the protection of the critically ill patients who participate in clinical research.
Conflicts of interestAuthors declared no conflicts of interest whatsoever.
Please cite this article as: Estella A. Ética de la investigación en el paciente crítico. Med Intensiva. 2018;42:247–254.

