


INTRODUCCION
La pancreatitis aguda (PA) es una enfermedad inflamatoria, en la que las formas de presentación grave cursan con necrosis digestiva del páncreas. Diferentes estudios realizados han investigado los estadios tempranos intracelulares que conducen a daño celular y a pancreatitis. La posibilidad de estudiar estas fases en el hombre es difícil, a la mayoría de los estudios están realizados experimentalmente con diferentes modelos animales, dado las dificultades que encierra investigar clínicamente la PA1.
Existen aspectos de esta enfermedad que están pendientes de ser aclarados, y aunque se ha avanzado en áreas, como los factores predisponentes, enfermedad y eventos bioquímicos dentro del páncreas, por el enorme número de estudios experimentales y clínicos, queda por conocer de forma más profunda los mecanismos de la puesta en marcha de la cascada de acontecimientos celulares.
La complejidad de la PA no se debe sólo a la variedad en las diferentes etiologías que se relacionan con la enfermedad, sino también al grado de gravedad, a la diferente forma de evolución y a otros factores, como:
1. La incidencia baja de formas graves que conlleva dificultad en el diseño de estudios aleatorizados en un centro regional en un período determinado.
2. Las diferencias entre los centros hospitalarios en relación al diagnóstico inicial, los conceptos terapéuticos y los intereses en la investigación, hacen difícil el diseño de un estudio en común.
3. La indicación de la cirugía temprana en la PA se ha hecho más restrictiva, y en consecuencia pocos pacientes con PA se trasladan a centros especializados; así los derivados son pacientes con PA muy grave que no pueden aleatorizarse en estudios con intervenciones terapéuticas tempranas.
4. El inicio de la enfermedad no puede determinarse con exactitud (habitualmente nos estamos refiriendo al inicio del dolor abdominal).
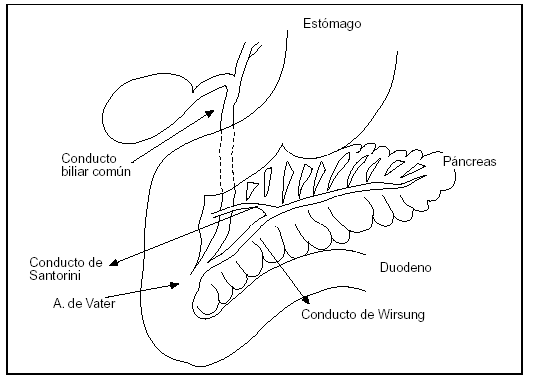
5. La localización del páncreas en el retroperitoneo hace inaccesible investigaciones detalladas (fig. 1).
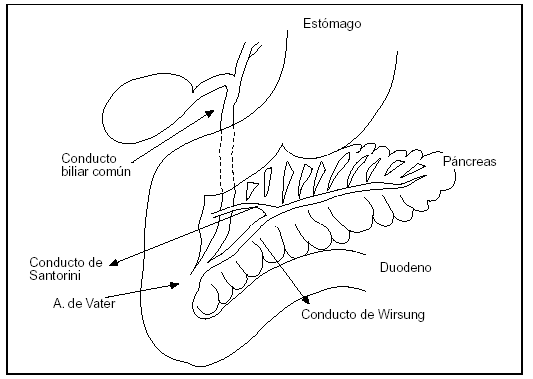
Figura 1. Páncreas situado en el marco duodenal.
6. Las PA graves requieren ingreso en unidades de cuidados intensivos (UCI) donde la coordinación de los recursos disponibles se realiza por el intensivista de forma interdisciplinaria; por ello la implicación de este especialista es imprescindible en España para la organización y la planificación en el diseño de estudios multicéntricos2.
LOS MODELOS ANIMALES EN LAS PANCREATITIS AGUDAS Y SU RELEVANCIA EN LA CLINICA
La investigación experimental con animales ha ayudado a comprender la fisiopatología de la PA; sin embargo, no ocurre lo mismo cuando se aplica el modelo animal en las investigaciones orientadas a las intervenciones terapéuticas. Un ejemplo sería la eficacia demostrada en modelos animales del antagonista de la colecistocinina y de los inhibidores de la proteasa, y el fracaso en su aplicación clínica3-5. Estas discrepancias pueden explicarse por las diferencias metodológicas. Si en el modelo animal las sustancias terapéuticas se aplicaron antes del desarrollo de la propia enfermedad, en la clínica se hizo pasadas 24-36 h del inicio de los síntomas; el número de pacientes incluidos en los ensayos clínicos era pequeño para obtener poder estadístico suficiente; finalmente, el modelo de reproducción de la PA no era similar al desarrollo de la PA a escala humana.
Foitzik T et al han especificado los puntos que cualquier modelo animal debe considerar para estudiar la PA2:
1. Pancreatitis necrótica grave.
a) Necrosis intra y extraparenquimatosa.
b) Signos de inflamación sistémica y disfunción orgánica (FMO).
c) Mortalidad reproducible (no tratados del 30 al 50%).
2. La fase del curso de la enfermedad.
a) Desde el inicio de la respuesta inflamatoria sistémica (SIRS) al daño pancreático con disfunción orgánica temprana (FMO).
b) Respuesta inflamatoria secundaria o exacerbación asociada a la infección pancreática con FMO tardía o séptica.
3. Respuesta a la terapia
a) Mejoría de la FMO temprana y reducción de la mortalidad temprana con la reposición hídrica y las medidas de soporte intensivo.
b) Reducción de las complicaciones infecciosas y de la mortalidad tardía con varias medidas (tratamiento antibiótico y estabilización de la barrera de la mucosa gastrointestinal).
4. Monitorización de parámetros fundamentales.
a) Protocolos que reproduzcan y verifiquen la gravedad de la pancreatitis.
b) Posibilidad de continuar o de repetir el registro de la funcionalidad de los órganos vitales durante la terapia y después de la terapia.
En los estudios terapéuticos más que la etiología tiene más valor la posibilidad de relacionar los problemas morfológicos con los síntomas sistémicos y la fase de la enfermedad. Por ello deben ser animales que permitan valorar la funcionalidad de los órganos vitales, el daño local o necrosis del páncreas y la mortalidad. Los estudios realizados en animales que correlacionaban en el pasado los parámetros analíticos (amilasa, lipasa, citocinas) con la gravedad de la enfermedad están hoy, en parte, en controversia.
La rata es, quizá, el animal más utilizado en los modelos experimentales de la PA; los ratones por su pequeño peso y por la dificultad para monitorizar la función orgánica no reúnen las condiciones ideales para las investigaciones terapéuticas. En estos casos serían ideales animales de mayor tamaño como cerdos, gatos, ovejas o perros, si bien las limitaciones éticas y el alto coste han dificultado en Europa el desarrollo de estudios con monitorización de parámetros orgánicos durante varios días.
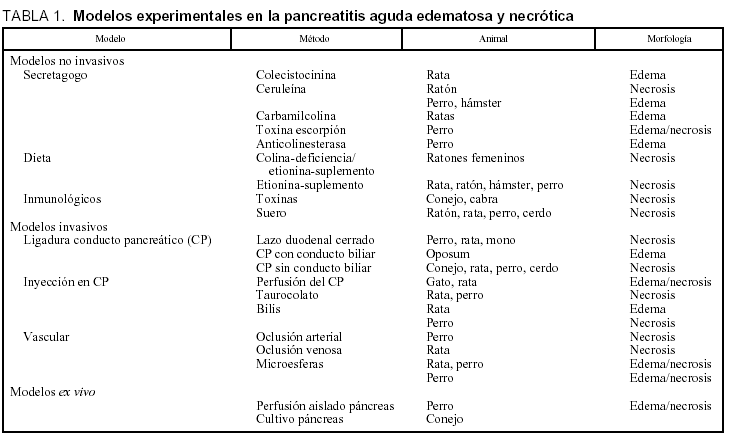
En la tabla 1 se exponen de forma resumida los diferentes modelos experimentales de la PA. Estos modelos se diferencias según a) la técnica de inducción de invasivos, no invasivos o ex vivo; b) la causa (biliar, obstructiva, alcohólica, tóxica o traumática, isquémica, etc.), o c) grado de gravedad (media/edematosa frente a grave/necrótica). Los modelos más ampliamente usados han sido revisados por diferentes autores en los últimos 10 años3,4,6-12.
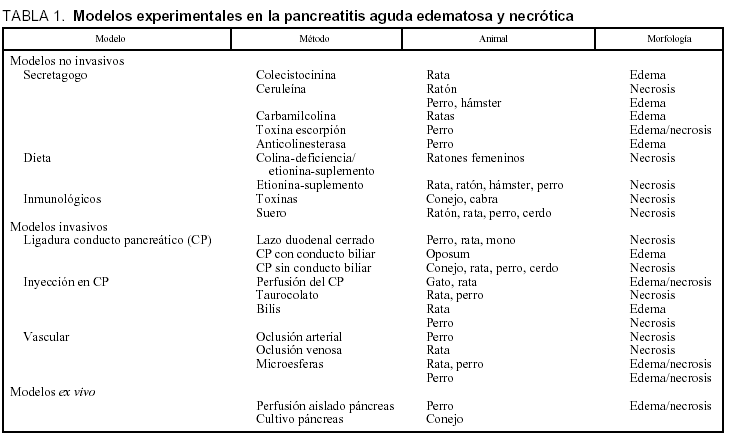
En el modelo no invasivo de secretagogos todas las sustancias utilizadas (colecistocinina; el análogo de la colecistocinina, como la ceruleina; agonistas colinérgicos, como la carbamicolina; el veneno del escorpión, y agentes anticolinesterasa) inducen PA por la excesiva estimulación del páncreas13-19. El modelo de la ceruleína se ha utilizado ampliamente para el análisis de los episodios intracelulares en la fase temprana de la PA, para relacionar el grado de lesión según la dosis utilizada y para el análisis de las lesiones pulmonares provocadas por la PA. Con 5-10 μg/kg/h en la rata se induce un incremento de enzimas en sangre asociado con edema intersticial pancreático, y el grado máximo de lesión a las 6-12 h tras la infusión de la ceruleína, con recuperación íntegra de la estructura pancreática. Sin embargo, este modelo tiene limitación de la no reproducción de formas más graves necróticas de PA.
En el modelo no invasivo de PA inducida por dieta por deficiencia de colina y/o suplemento de etionina se reproduce un modelo con cierta similitud al humano por el desarrollo hacia la necrosis de forma progresiva (la mortalidad puede variar desde el 0 al 100% según la cantidad de dieta administrada), la respuesta bioquímica y los cambios histológicos. Sin embargo el modelo experimental al ser con animales de pequeño tamaño conlleva dificultades para la investigación de sustancias terapéuticas en la PA 20-22.
El modelo inducido por la respuesta inmunológica a toxinas o sueros en la PA se basa en la inyección de toxinas de meningococo o Escherichia coli en el conducto pancreático del conejo, seguido de la misma sustancia diluida al 1/40 por vía intravenosa. En un período variable de 4-24 h se desarrolla una PA necrótica con una mortalidad del 100%. La sensibilización inmunológica se ha reproducido también con 1 g de ovalbúmina intravenosa seguido de 200 mg subcutáneos al quinto día. A las 4 semanas una inyección intraductal de ovalbúmina induce una PA intersticial o edematosa y a las 8 semanas otra favorece el desarrollo hacia la forma necrótica. Este modelo se ha desarrollado escasamente por la dificultad de regular la graduación de las lesiones y por la alta mortalidad que impide estudiar de forma más detenida la patogenia23,24.
El modelo invasivo del lazo cerrado del duodeno fue de los primeros modelos experimentales en PA descritos por Pfeffer et al en 1957. Se ligan los primeros 10 cm del duodeno, por debajo del píloro, y el conducto común biliar. El conducto pancreático se comunica con el duodeno ligado. Una gastroduodenostomía restablece el flujo gástrico. A las 4 h aparece edema de la cabeza y cuerpo del páncreas. Entre las 9 y 12 h se aprecia una PA hemorrágica sin necrosis grasa. El modelo de PA incluye distensión del duodeno ligado con reflujo de las enzimas activadas. A este modelo se le han añadido modificaciones como la ligadura del conducto pancreático y la inyección de bilis infectada en el duodeno ligado, junto con otras más. Se ha utilizado para el estudio etiológico de la PA, por la graduación de la gravedad en el estudio de diferentes sustancias terapéuticas y ha ayudado a profundizar en los factores patogénicos responsables del inicio de la PA, principalmente el papel del reflujo duodenal25-27.
La ligadura del conducto pancreático fue descrita por Opie et al en 1901 al describir dos pacientes que fallecieron por pancreatitis necrótica y el hallazgo de dos piedras impactadas en la papila de Vater, sugiriendo que este acontecimiento favorecería el paso de bilis al conducto pancreático con el inicio de PA. Esta teoría del canal común ha sido estudiada en diferentes modelos animales sobre todo en aquellos cuya anatomía del conducto pancreático y biliar era similar al humano, como es el oposum, provocando la ligadura del conducto común una PA con una mortalidad del 100% en el día 14. Este modelo se relaciona con la PA desarrollada con presencia de litiasis biliar en el canal común al provocar una obstrucción transitoria y un reflujo biliar al conducto pancreático. Sin embargo, Lerch et al demostraron que el grado de necrosis no se modificaba en diferentes variantes de este modelo: obstrucción del canal común, obstrucción por separado del conducto biliar y del pancreático y sólo del conducto pancreático28-30.
El modelo a baja presión de perfusión o de inyección de sustancias en el conducto pancreático reproduce la hipótesis del factor del reflujo biliar en la PA desencadenada por litiasis biliar. Este modelo permite modular la inducción de las lesiones inflamatorias a través del grado de presión en el líquido a perfundir, la concentración de las sustancias inyectadas y del tiempo de exposición. La PA edematosa puede ser inducida en gatos con diferentes sustancias ajustándose a diferentes modelos, en todos se perfunde en segundo lugar secreción pancreática activada; en el modelo biliar en primer lugar es una sal biliar (ácido glucodeoxicólico); en el modelo alcohólico se inyecta alcohol; en el modelo por hipercalcemia se perfunde por vía intravenosa gluconato cálcico, y por último, en el modelo de obstrucción-secreción tras obstruir el conducto principal pancreático en un 50% se perfunden sustancias como secretina y colescistocinina intravenosas que estimulan el páncreas. Cualquiera de los cuatro modelos anteriores se convierte en forma necrótica al añadir una perfusión de 16-dimetilprostaglandina E2. Este modelo en general es valorado para la patogenia de la PA y las diferentes modalidades terapéuticas, así como para la infección de la necrosis pancreática. Schmidt et al reproducen la PA necrótica con diferentes grados de gravedad, añadiendo a la infusión de ceruleína, con una presión y tiempo determinados, una inyección de ácido glucodeoxicólico, a diferente concentración y tiempo de infusión, dentro del conducto pancreático de las ratas. Las ventajas de este modelo de baja presión de perfusión son su similitud con las PA en el hombre con baja morbilidad y mortalidad, con un importante paralelismo en las etiologías, el tiempo de evolución y en los cambios histológicos31-35.
Ya Panum en 1986 informó de la PA hemorrágica focal por compromiso en la circulación del páncreas. Desde entonces, el modelo de PA inducida por cambios vasculares se ha usado de forma específica para estudiar los cambios fisiopatológicos, bien por problemas en la entrada o en la salida del flujo circulatorio, o por problemas en la microcirculación del páncreas. En este modelo se incluyen las PA desarrolladas en el contexto de un shock hipovolémico. Para que se produzcan cambios significativos histológicos en forma de necrosis es necesaria la obstrucción permanente de la arteria duodenopancreática superior. A escala venosa es necesaria la ligadura de la vena esplécnica y de la pancreaticoduodenal superior para que se produzcan cambios significativos en el páncreas. El modelo de la microcirculación se reproduce por la inyección de microesferas de poliestereno de 10 a 40 μm en la arteria pancreaticoduodenal superior de la rata favoreciendo la aparición de edema, infiltración inflamatoria celular, hemorragia y necrosis del páncreas. El modelo de la microcirculación tiene vigencia hoy por su similitud con situaciones de deplección de volumen, vasoconstricción química provocada por fármacos, cuagulación intravascular y aumento de la permeabilidad endotelial. El modelo vascular de acceso o de salida de los vasos principales pancreáticos tiene su correspondencia sólo con casos de arteritis, esclerosis arteriolar o trombosis de venas como la porta y/o esplécnica, a esto se le añade la necesidad de realizarlo con animales de tamaño grande lo que encarece la experiencia36-42.
El modelo ex vivo tiene aún escasa relevancia para la implicación clínica en el hombre43,44.
FACTORES ASOCIADOS A LA PANCREATITIS AGUDA. INCIDENCIA
Aunque los acontecimientos celulares son la base del desarrollo de la PA están descritos varios factores y enfermedades asociados al desarrollo de la pancreatitis aguda (tabla 2)45. De los modelos descritos experimentales, los más usados, para inducir la PA en modelos animales, tales como la perfusión de ceruleína o la dieta con deficiencia de colina y/o suplemento de etionina, no son reconocidos como causas de PA humana. Los cálculos biliares y el abuso del etanol que juntos explican cerca del 80% de los casos46, en ninguno de los modelos animales descritos anteriormente se duplican estas situaciones; sin embargo, la estructura y los cambios bioquímicos observados en la fase temprana de las PA, son constantes en los diferentes modelos animales, y cambios similares se han observado en la PA humana. La frecuencia relativa con la que cada una de estas etiologías descritas se encuentra depende en gran parte de la población que es evaluada. En Inglaterra algunos investigadores han publicado que dos tercios de sus pacientes tienen la litiasis biliar como causa, sobre todo entre mujeres y en el ámbito rural47. En los EE.UU. la principal causa es el alcohol, sobre todo entre varones y grandes ciudades48. Un 10% está causado por diversas causas, como hiperlipemia, la infección viral, diferentes fármacos, hipercalcemia, la obstrucción ductal y la perfusión pancreática deteriorada. Finalmente, aproximadamente el 10% de los casos son por causas idiopáticas49.

En España la incidencia se sitúa entre 30 y 50 casos por 100.000 habitantes/año y afecta especialmente a mujeres de mediana edad (alrededor de 55 años) en una proporción doble que a varones y relacionado con litiasis biliar. En los varones, además se relaciona con el abuso de alcohol50.
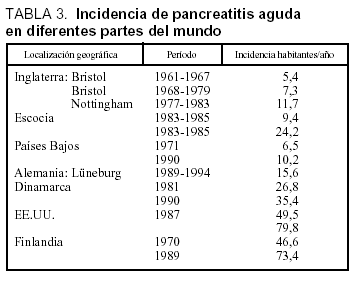
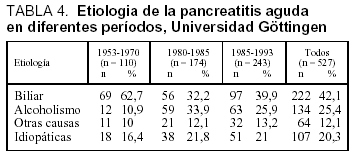
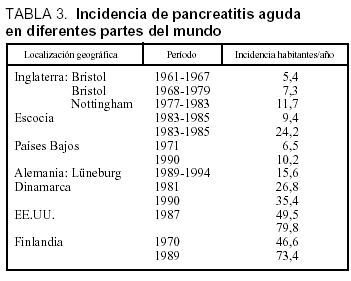
En la tabla 3 está desarrollada la incidencia de la PA en diferentes partes del mundo, apreciándose una tendencia al ascenso y en probable relación con los hábitos alimentarios51. Estudios realizados en la misma área geográfica muestran un cambio en la etiología, reduciéndose la causa biliar y ascendiendo la etílica, este ascenso en las cifras de incidencia puede estar relacionado con los hábitos alimentarios (tabla 4)52-53.
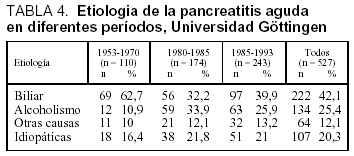
En relación con el género, Lankish et al51 estudian 18 publicaciones en el período de 1949 a 1994 con más de 100 pacientes, valorando el género y la etiología de la primera crisis de PA. De los 6.749 pacientes estudiados el 57,4% eran varones y el 42,6% mujeres; en el 38,1% la etiología era biliar y en el 35,4% alcohólica. La mayor incidencia global en el varón puede explicarse por la suma de las dos etiologías principales de la PA: la biliar y la etílica.
La colangiopancreatografía retrógrada (CPRE) desempeña un papel fundamental en la evaluación de pacientes con PA idiopática y se debe realizar después de la recuperación del paciente del episodio agudo54. Aunque la ecografía abdominal es muy sensible en la detección de litiasis en la vesícula biliar, un 4-7% de los casos pueden ser no detectados55, y en la litiasis del conducto biliar común la ecografia sólo es sensible en el 50-75% de los pacientes56,57. Además, la PA se asocia normalmente con piedras pequeñas (< 3 mm), siendo más difícil el diagnóstico por ultrasonografía58. Recientemente, dos estudios prospectivos de pacientes consecutivos con PA idiopática, entre dos tercios y un 75% de los casos, tras estudios iniciales, encontraron microlitiasis mediante el examen microscópico de la bilis, en forma de cristales de monohidrato de colesterol o gránulos de bilirrubinato cálcico59,60.
LA PANCREATITIS EMPIEZA EN LAS CÉLULAS ACINARES
A pesar de las limitaciones del modelo animal, los datos sugieren que, con independencia del episodio o mecanismo inicial, una cascada similar de acontecimientos ocurre una vez que la pancreatitis se inicia. Los mecanismos moleculares a escala celular que envuelven la patogenia de la PA están escasamente clarificados. Diversas situaciones pueden precipitar la PA en humanos, aunque solamente una pequeña proporción de pacientes con estos factores precipitantes desarrolla la enfermedad: en un 3 a un 7% de los casos de litiasis biliar, en un 10% de los alcohólicos y en unos pocos casos con hipercalcemia61,62.
El mecanismo exacto de inducción de la PA por estos agentes no es conocido, aunque ya se ha descrito que el modelo animal a baja presión de perfusión o de inyección de sustancias en el conducto pancreático, junto con el lazo del duodeno, reproducen la hipótesis del factor del reflujo biliar en la PA desencadenada por litiasis biliar, y la ligadura del conducto pancreático o la teoría del canal común a la PA con presencia de litiasis biliar en dicho canal28,30. En la hiperlipemia, la concentración tóxica de ácidos grasos libres liberados desde los triglicéridos por la acción de la lipasa pancreática en los capilares pancreáticos63. Sin embargo, no está claro por qué la pancreatitis inducida por alcohol ocurre sólo después de algunos años y no después de una gran ingestión tóxica.
Un importante número de investigadores han intentado definir los episodios celulares que están en el desarrollo de la PA. En condiciones normales, diferentes mecanismos naturales protegen al páncreas de la autodigestión por las proteasas pancreáticas. Estos mecanismos son46:
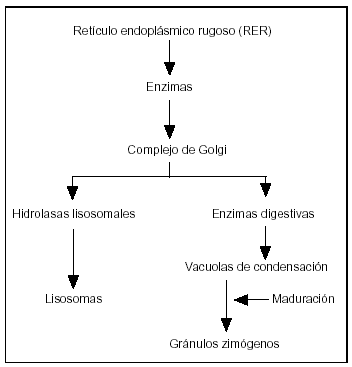
1. Las enzimas pancreáticas se sintetizan en el retículo endoplásmico de la célula acinar, se almacenan en vacuolas que al madurar se transforman en zimógenos, los cuales cuando se fusionan en la membrana celular se liberan en la luz acinar (fig. 2). La activación de estas proenzimas (p.ej., tripsinógeno, proelastasa) normalmente ocurre en el duodeno, donde la enteropeptidasa secretada por la mucosa duodenal activa el tripsinógeno a tripsina, que activa las proenzimas restantes.
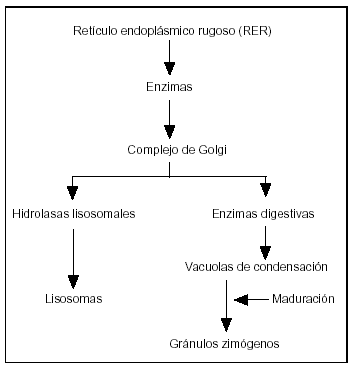
Figura 2. La fisiología secretora de la célula acinar pancreática empieza con la síntesis de las enzimas en el retículo endoplásmico. Las enzimas son transportadas al aparato de Golgi. Las hidrolasas lisosomales son segregadas dentro de los lisosomas y las enzimas digestivas en unas vacuolas de condensación, madurando en gránulos de zimógenos, que son descargados dentro de la luz acinar fusionándose con la membrana celular.
2. La presencia de inhibidores de la tripsina del páncreas puede inactivar el 20% de la actividad de la tripsina; una segunda línea de defensa es la mesotripsina, la enzima de: la tripsina en sí misma, lisa e inactiva la tripsina, y las antiproteasas circulantes no específicas (α1-antitripsina y α-2-macroglobulina) previenen la activación de proenzimas pancreáticas dentro del tejido pancreático o de sus conductos. Cantidades más grandes liberadas de tripsina, sin embargo, conllevan insuficiencia de los mecanismos de defensa del páncreas y del sistema en su conjunto, activando otras enzimas.
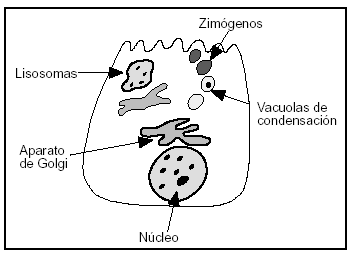
3. En el retículo endoplásmico de la célula acinar se segregan las enzimas digestivas, posteriormente son transportadas al aparato de Golgi, de donde son liberadas en unas vacuolas condensativas que al madurar se convierten en zimógenos, liberándose posteriormente en la luz acinar (figs. 2 y 3). En la célula normal los zimógenos y las enzimas lisosomales o hidrolasas, como la captesina B, se segregan de forma separada de la red del aparato de Golgi impidiendo su contacto.
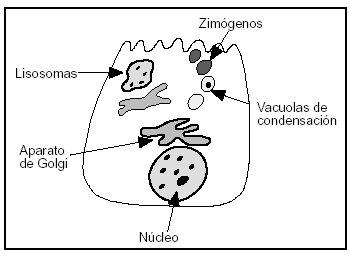
Figura 3. Ilustración de la estructura de la célula acinar del páncreas.
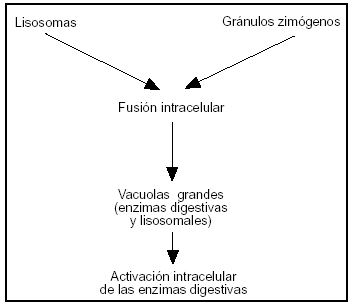
Los estudios más recientes sugieren que una anomalía en el transporte y secreción en la célula acinar de las proteínas enzimáticas juegan un importante papel en la evolución de la PA64,65. Modelos experimentales de PA demostraron unión de gránulos de zimógeno con vacuolas del lisosoma, fenómeno denominado "colocalización" o "crinofagia" (fig. 4). La activación prematura del tripsinógeno por la catepsina, enzima lisosomial, es el primer acontecimiento66,67; posteriormente estas vacuolas del citoplasma se vuelven más inestables, favoreciéndose la liberación de enzimas, proteolíticas y lipolíticas, en el citoplasma de la célula y la cascada de activaciones enzimáticas (fig. 5).
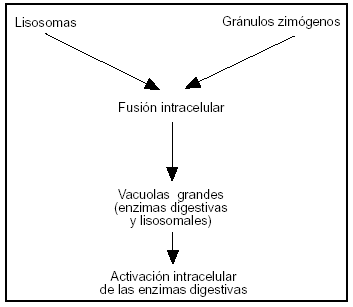
Figura 4. Fusión intracelular de los gránulos de zimógenos con lisosomas (colocalización o crinofagia) permitiendo la posible activación de los zimógenos por las hidrolasas. Estas organelas conteniendo los zimógenos digestivos y las hidrolasas lisosomales se debilitan y pueden liberar las enzimas en el citoplasma.
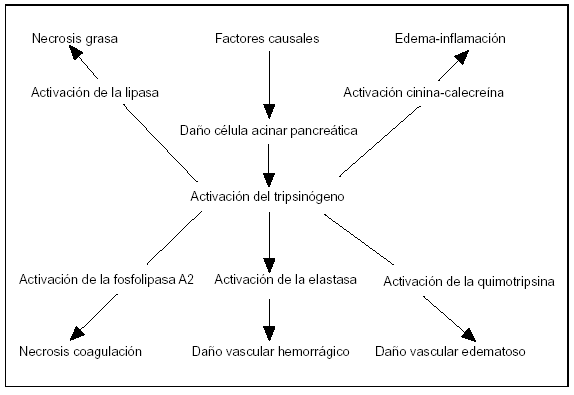
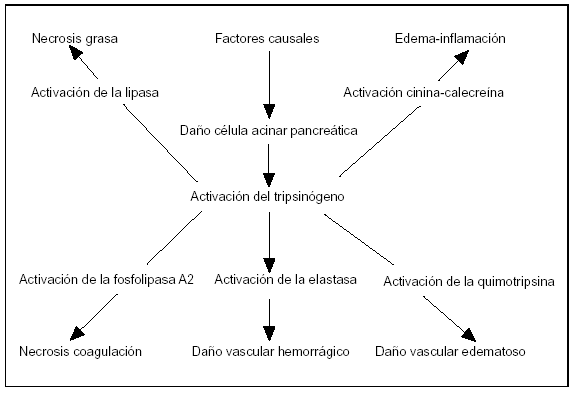
Figura 5. La activación del tripsinógeno en la patogenia de la pancreatitis aguda.
Esto va acompañado por un aumento de la concentración de calcio intracelular, que se libera del retículo endoplásmico rugoso. Este incremento de (Ca+) va seguido de deshidratación de la célula y bloqueo en la secreción de proteínas en los conductos acinares68. El desarrollo de estos tres factores da por resultado la activación del tripsinógeno dentro de la célula acinar64, dando lugar al proceso de la autodigestión69.
El calcio liberado intracelular no sólo es mediador del daño acinar, sino que tambien regula las actividades de numerosas enzimas, hormonas y factores de crecimiento, participando directamente en la patogenia de la PA70,71.
La activación del tripsinógeno está considerada como un escalón fundamental en la evolución de la activación de la PA. Una vez la tripsina se ha activado por el tripsinógeno, su inhibición no altera el curso de la pancreatitis, ya que otras proteasas continúan con el daño celular. Esto explica por qué los inhibidores de la tripsina no alteran el curso de la PA. La activación de las enzimas pancreáticas en la célula acinar antes de ser secretadas en la luz acinar es una de las teorías de la patogenia de la PA72.
El potencial nocivo de las enzimas digestivas en las células acinares del páncreas es notablemente diferente73. La elastasa, la lipasa, la quimotripsina y la fosfolipasa-A2 son más potentes al dañar las células acinares cuando se comparan con la tripsina molecular (fig. 5). Aunque la activación del tripsinógeno comienza la activación y desarrolla la respuesta en cascada inflamatoria, esta enzima es la que menor daño directo realiza al páncreas.
PATOGENIA DE LA PANCREATITIS HEREDITARIA
La prematura activación de los zimógenos pancreáticos se ha propuesto como mecanismo patogénetico en las crisis de PA con base hereditaria con mutación en el brazo largo del cromosoma 7(7q35) y en el gen del tripsinógeno catiónico en el residuo R117, resultando un fallo del mecanismo de seguridad de la célula acinar y activándose la tripsina74-76. Sin embargo, no todos los pacientes con pancreatitis hereditaria tienen una mutación a nivel del R117, y no todos los pacientes con la mutación en el gen del tripsinógeno desarrollan la enfermedad, un 20% no la padecen.
Por otro lado se ha demostrado en pacientes con brotes de PA recurrente, con páncreas divisum o fibrosis quística, una mutación en al menos un alelo de la fibrosis quística conductasa transmebrana reguladora (FQCTR). Esta mutación está asociada a la mayor producción y concentración de la secreción pancreática y conlleva una obstrucción ductal y una alteración de la célula acinar, reduciéndose el pH y provocando un transporte anómalo intracelular77,78.
LA MICROCIRCULACION EN LA PANCREATITIS AGUDA
La liberación de las enzimas pancreáticas daña el endotelio vascular así como las células acinares79-81. Los cambios en la microcirculación en una fase temprana de las PA pueden favorecer el desarrollo de la enfermedad. Posibles mecanismos como el aumento de la permeabilidad capilar, la reducción del flujo sanguíneo, la interacción de los leucocitos del endotelio vascular, la formación de trombos, la vasoconstricción, la estasis capilar, el descenso de la saturación de oxígeno y la progresiva isquemia, ocurren de forma temprana en los modelos animales de PA. Estos cambios conllevan la extravasación de líquido en el espacio perilobular y la inflamación de la glándula (pancreatitis edematosa e intersticial).
Existe especulación sobre el papel de la isquemia-reperfusión en el daño del páncreas81, aunque cada vez se reconoce más en la patogenia el mecanismo de isquemia/reperfusión, sobre todo cuando nos referimos al paso de pancreatitis edematosa a necrótica. El mecanismo de este daño está bien establecido en el corazón, músculo esquelético e intestinos. El daño tisular secundario a la reperfusión conlleva liberación de radicales libres de citocinas inflamatorias en la circulación.
Estudios con leucocitos marcados con radionucleótidos como el indio-111 han demostrado invasión del páncreas por macrófagos y leucocitos polimorfonucleares en los estadios iniciales de la PA experimental y en humanos82-84. La activación del complemento y la liberación del C5a demuestra un papel significativo en la respuesta de estas células inflamatorias. Los granulocitos y los macrófagos activados causan liberación de citoquinas proinflamatorias (factor de necrosis tumoral, interleucinas 1, 6 y 8), los metabolitos del ácido araquidónico (prostaglandinas, factor de activador de plaquetas y leucotrienos), enzimas lipolíticas y proteolíticas, de radicales libres de oxígeno, siendo soprepasada la capacidad antioxidante del sistema endógeno, y aumento de la concentración de calcio intracelular. Estas sustancias también interactúan con la microcirculación pancreática aumentando la permeabilidad e induciendo trombosis y hemorragia; así la PA evoluciona hacia la necrosis, resultando un círculo vicioso en el que se liberan más enzimas pancreáticas y ocurre más daño pancreático, propiamente dicho85-89. Los radicales libres de oxigeno son unos potentes agentes oxidantes que a través de la peroxidación lesionan los componentes lipídicos mitocondriales y de la membrana celular, lesionando el endotelio y favoreciendo la permeabilidad capilar87. La administración de catalasa y de dismutasa experimental antes de la inducción de PA, disminuye el daño del tejido pancreático90; no obstante, la administración tras estar establecida la PA evidencia sólo un limitado beneficio91.
La adhesión de los leucocitos a los vasos en la microcirculación es un episodio secundario a los cambios de la permeabilidad capilar de la PA.
Las enzimas pancreáticas activadas, la disfunción de la microcirculación y la liberación de mediadores inflamatorios conlleva un rápido deterioro del tejido pancreático y necrosis. La interacción entre estos factores hace difícil valorar el papel desempeñado individualmente por ellos para inducir la lesión pancreática. Los factores que inciden en limitar el daño pancreático no se comprenden bien, haciendo que la pancreatitis aguda no evolucione hacia necrótica en un 80%.
LA RESPUESTA SISTÉMICA EN LA PANCREATITIS AGUDA
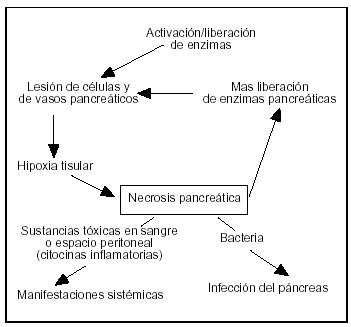
Una vez que se activa la célula acinar sigue una cascada imprevisible de episodios que puede llevar localmente a la inflamación del intersticio o a necrosis grave, extendiéndose a los espacios peripancreáticos y a la secreción de factores tóxicos en la circulación sistémica o espacio peritoneal, induciendo manifestaciones sistémicas o fracaso de los diferentes órganos secundarios a la PA (fig. 6).
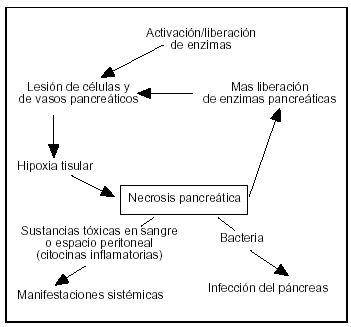
Figura 6. Durante la pancreatitis aguda, junto con los episodios incipientes, varias citocinas inflamatorias son producidas, tanto a escala local como sistémica. La cascada se desarrolla y se amplía rápidamente envolviendo varios mediadores; cada uno de ésos tiene un papel en el desarrollo de las manifestaciones sistémicas. El desarrollo de la necrosis pancreática infectada tiene un factor pronóstico adverso.
La secreción de tripsina activa el complemento y el sistema de cinina-calecreína92,93. Los productos del sistema calecreina-cinina causan vasodilatación, aumento de la permeabilidad vascular y acumulación de leucocitos, favoreciendo la inestabilidad hemodinámica93. Concentraciones elevadas sistémicas de estos péptidos vasoactivos pueden contribuir a trastornos hemodinámicos, a cuagulación intravascular diseminada, al shock, insuficiencia renal y al síndrome de fallo multiorgánico92.
La tripsina activa la fosfolipasa-A2, esta enzima en presencia de los ácidos biliares destruye las membranas celulares, causando necrosis del parénquima pancreático. Si bien hay inhibidores para la tripsina y otras enzimas proteolíticas, no existe para la fosfolipasa-A2; ésta y otros tóxicos de la membrana lisofosfolipídica celular alcanzan la circulación y causan daño al surfactante pulmonar, y son responsables del síndrome de distrés respiratorio94.
En las pancreatitis clínicas y experimentales, la fosfolipasa-A2 sérica se correlaciona con la gravedad de la enfermedad95,96.
La elastasa es bastante perjudicial por su efecto directo celular, lesiona la elastina de los vasos, produciendo complicaciones hemorrágicas siendo su espectro de acción amplio, en numerosas proteínas73. Estudios recientes sugieren que la liberación de la elastasa conlleva la activación de las células inflamatorias o neutrófilos 37 con un importante papel citotóxico a nivel de órganos distantes del páncreas.
La lipasa, sintetizada en las células acinares en forma activa, necesita de la presencia de ácidos biliares para su actividad biológica, su liberación en el espacio peripancreático induce necrosis de la grasa. Otros sistemas en cascada activados en la PA son la cuagulación-fibrinólisis y el sistema del complemento97,98. Estas dos activaciones junto con la cascada de la calecreína-cinina tienen importancia en la fisiopatología de la PA, contribuyendo a las complicaciones por la toxicidad sistémica98. La respuesta inflamatoria que se extiende de forma generalizada es responsable de la morbilidad y mortalidad en la PA99.
La mayoría de las investigaciones en la actualidad están centradas en identificar los inductores de esta enfermedad sistémica, destacando diferentes citocinas (IL) como IL-1, IL-6, IL-8, factor de necrosis tumoral (TNF) y factor activador plaquetario. Estas citocinas se consideran los principales mediadores en la transformación de la PA de un proceso local inflamatorio a una enfermedad multiorgánica.
Además de la hipercitocinemia resultante del proceso inflamatorio local pancreático y de los tejidos peripancreáticos, existe evidencia de que son liberadas también por los macrófagos hiperactivados cuando la pancreatitis se complica con infección; estas citocinas activan los neutrófilos que han infiltrado órganos vitales, como el pulmón, el hígado y los órganos digestivos, y se realiza un "segundo ataque"100,101. Los neutrófilos a través de las enzimas proteolíticas y oxidantes lesionan los órganos vitales infiltrados, causando daño celular y disfunción de los órganos distales al páncreas.
Una explicación en la patogenia de la PA con estos mediadores inflamatorios ha llevado a plantear diferentes preguntas en relación al tratamiento de esta enfermedad. Aunque a escala experimental han sido relevantes los resultados usando antagonistas de las citocinas, la estrecha ventana terapéutica en pacientes sépticos con los antagonistas específicos de las citocinas hace enormemente complejo su uso en la práctica clínica, incluso con el antagonista del factor activador plaquetario, lexipafant102. En el futuro las líneas de investigación desarrollaran terapias específicas para inhibir la progresión destructiva de la glándula y el desarrollo de complicaciones sistémicas.
LA LESION PULMONAR EN LA PANCREATITIS AGUDA
Aproximadamente un tercio de los pacientes con PA desarrollarán lesión aguda pulmonar (LAP) y síndrome del distrés respiratorio del adulto (SDRA)103, con una mortalidad del 60% en la primera semana103. La excesiva citotoxicidad de los neutrófilos parece estar implicada en el desarrollo del daño pulmonar104. El aumento de la permeabilidad microvascular conlleva liberación al espacio alveolar de un trasudado rico en proteínas con una disminución de la distensibilidad pulmonar. La activación de la fosfolipasa A favorece la digestión de la lecitina, el mayor componente del surfactante pulmonar. Clínicamente aparece una hipoxemia con los síntomas acompañantes de infiltrado bilateral difuso. En modelos de LAP experimental se induce edema intraalveolar, reducción de la vía aérea distal105, daño en las células endoteliales, y secuestro de los leucocitos106 evidenciándose la actividad de la mieloperoxidasa107, 108.
El papel de los neutrófilos (reclutamiento, adhesión y activación) y la circulación de los mismos, la activación de las vías de señales de transducción y las deficiencias en la producción de surfactante pulmonar por los neumocitos tipo II109 son claves en las complicaciones a distancia del páncreas no sólo en el pulmón sino también en el colon, el hígado y los riñones, al provocar trastornos en la microcirculación en estos órganos.
La activación de los neutrófilos conlleva una respuesta del sistema inmunológico del huésped, que si es inadecuada puede amortiguarse con intervenciones que modulen esta citotoxicidad de los neutrófilos, como se ha demostrado con diferentes estudios110,111. Esta activación se induce por la activación del complemento, la producción de las citocinas y la estimulación de la expresión de la molécula de adherencia y de los macrófagos alveolares112. La secreción de sustancias quimioatrayentes como la IL-8, el TNF, la βIL-1, fMet-Leu-Phe (un producto de la pared bacteriana) y el factor del complemento C5a, da como resultado la concentración de neutrófilos en la microcirculación pulmonar. La exposición de los neutrófilos a los mediadores inflamatorios endógenos conlleva una disregulación de la expresión de la β2 integrina y a reforzar el potencial adhesivo.
Se ha encontrado correlación entre la gravedad de la PA y la activación113, y la concentración del TNF, y la IL-8 tanto en suero114 como en el líquido ascítico115 y en el líquido alveolar116. La IL-8 es quimioatrayente y activadora de los leucocitos T114; la IL-8 se encontró en los espacios aéreos de pacientes con SDRA y se asocia con mortalidad elevada117. Otro estudio reciente ha encontrado en una serie de 11 pacientes con PA una elevación de IL-8, interleucina-6 y expresión neutrófila CD11b significativamente más elevada en pacientes que desarrollaron ALI118.
Hartwing et al han desarrollado nuevas técnicas con análogos de la glucosa, fluordioxiglucosa (FDG), en un modelo experimental con ratas, permitiendo medir la actividad de los neutrófilos en el pulmón en las PA y favoreciendo el conocimiento de la compleja fisiopatología de la LAP y la PA119. Los futuros estudios deberan confirmar en seres humanos la sensibilidad, la especificidad y el valor predictivo de esta técnica en el diagnóstico y valoración de la gravedad de la respuesta inflamatoria de la PA en el pulmón120.
CONCLUSION
La investigación en la PA experimental nos ha permitido conocer el inicio y el desarrollo de la enfermedad en la célula acinar, en el tejido pancreático, peripancreático y a escala sistémica, con la afectación de órganos que inciden en el aumento de la mortalidad como es la activación de los neutrófilos en el pulmón.
El conocimiento de la fisiopatología en la PA nos revela la importancia de un diagnóstico temprano con medidas, fáciles y en la cabecera del paciente, de marcadores biológicos que nos ayuden a detectar a pacientes potencialmente graves, indicando su ingreso en las unidades de cuidados intensivos para favorecer un soporte (hemodinámico y respiratorio) adecuado y como medidas que contrarresten la situación patológica de un páncreas con afectación celular, tisular y en su microcirculación. La modulación de la respuesta inflamatoria con medidas de intervención terapeútica específicas que impidan la disfunción orgánica, frenen el proceso de destrucción glandular y favorezcan la regeneración de la misma, son aspectos pendientes que tienen una ventana de actuación determinada y donde el retraso en dichas actuaciones hará el proceso irreversible.

