







Edited by: Federico Gordo - Medicina Intensiva del Hospital Universitario del Henares (Coslada-Madrid)
Last update: December 2023
More infoEnterobacterales resistant to carbapenems or producing extended-spectrum β-lactamases (ESBL) and non-fermenters resistant to carbapenems present resistance to many of the antimicrobials commonly used in clinical practice, and have been recognized by the World Health Organization as a critical priority for the development of new antimicrobials. In this review, the main mechanisms of resistance of Enterobacterales, Pseudomonas aeruginosa, Acinetobacter baumannii and Stenotrophomonas maltophilia to β-lactams, quinolones, aminoglycosides and polymyxins will be addressed. Updated information will be presented on the importance in resistance of antimicrobial modification mechanisms (including class C or extended-spectrum β-lactamases, carbapenemases and aminoglycoside-modifying enzymes), permeability alterations due to porin or lipopolysaccharide expression disorders, production of active efflux pumps, target alterations or protection, and expression of two-component systems.
Los Enterobacterales resistentes a carbapenémicos o productores de betalactamasas de espectro extendido (BLEE) y los no fermentadores resistentes a carbapenémicos presentan resistencia a muchos de los antimicrobianos comúnmente empleados en la práctica clínica, y han sido reconocidos por la Organización Mundial de la Salud como una prioridad crítica para el desarrollo de nuevos antimicrobianos. En esta revisión se abordarán los principales mecanismos de resistencia de los Enterobacterales, Pseudomonas aeruginosa, Acinetobacter baumannii y Stenotrophomonas maltophilia a betalactámicos, quinolonas, aminoglucósidos y polimixinas. Se presentará información actualizada sobre la importancia en la resistencia de mecanismos de modificación de antimicrobianos (incluyendo betalactamasas de clase C de espectro extendido, carbapenemasas y enzimas modificadoras de aminoglucósidos), alteraciones de la permeabilidad por trastornos en la expresión de porinas o del lipopolisacárido, producción de bombas de expulsión activa, alteraciones de la diana o protección de la misma y expresión de sistemas de doble componente.
Resistance to antimicrobials is one of the main healthcare problems worldwide, particularly in relation to multiresistant microorganisms. The World Health Organization (WHO) has published a list of priorities referred to bacteria for which the development of new antimicrobials is an urgent need. In this context, a critical concern refers to Enterobacterales resistant to carbapenems or producing extended-spectrum beta-lactamase (ESBL) and non-fermenting species (Pseudomonas aeruginosa, Acinetobacter baumannii) resistant to carbapenems.1 Moreover, these microorganisms usually present resistance to other groups/families of antimicrobials commonly used in clinical practice. The present study provides a succinct review of the principal mechanisms of resistance of Enterobacterales, P. aeruginosa, A. baumannii and also Stenotrophomonas maltophilia.
EnterobacteralesResistance to betalactamsThe principal mechanism of resistance to beta-lactams in Enterobacterales is the production of beta-lactamases – enzymes that hydrolyze the beta-lactam ring, inhibiting its antibacterial activity.2,3 Four classes of beta-lactamases have been established, based on their molecular structure (A, B, C and D).4 The enzyme activity depends on a serine group in classes A, C and D, and on one or two zinc ions in class B; as a result, the latter are also known as metallo-β-lactamases.5 On the other hand, the beta-lactamases can be classified into three functional groups (1, 2 and 3) according to their capacity to hydrolyze different substrates and their inhibition by different compounds (Table 1).6,7 From the clinical perspective, the enzymes of greatest interest in enterobacteria correspond to three groups: ESBL, class C enzymes and carbapenemases.2,3
Classification of beta-lactamases of clinical interest.
| Functional groupa | Molecular class | Reference substrates | Inhibition | Type of enzyme | Examples (families/representatives) | |
|---|---|---|---|---|---|---|
| AC-TZB | EDTA | |||||
| 1 | C | Cephalosporins | − | − | Chromosomal cephalosporinases Plasmidic cephamycinases | AmpCsFOX, DHA, ACT… |
| 2a | A | Penicillins | + | − | Penicillinases | PC1 |
| 2b | A | Penicillins Cephalosporins-1 G | + | − | Broad-spectrum penicillinases | TEM-1, SHV-1 |
| 2be | A | Cephalosporins-ES Monobactams | + | − | ESBL | TEM, SHV, CTX-M… |
| 2br | A | Penicillins | − | − | TEM resistant to inhibitors | TEM-30… |
| 2ber | A | Cephalosporins-ES Monobactams | − | − | ESBL resistant to inhibitors | TEM-50… |
| 2c | A | Carbenicillin | + | − | Penicillinases | PSE-1 |
| 2ce | A | Carbenicillin Cefepime | + | − | Penicillinases -cefepimases | RTG-4 |
| 2d | D | Oxacillin | ± | − | Oxacillinases | OXA-1, OXA-10… |
| 2de | D | Cephalosporins-ES | ± | − | Extended-spectrum oxacillinases | OXA-11… |
| 2df | D | Carbapenems | ± | − | OXA type carbapenemases | OXA-23, OXA-48… |
| 2e | A | Cephalosporins-ES | + | − | Carbapenemases | CepA |
| 2f | A | Carbapenems | ± | − | Carbapenemases | KPC, IMI, GES-6… |
| 3 | B | Carbapenems | − | + | Carbapenemases | IMP, VIM, NDM…L1, CphA… |
AC/TZB: clavulanic acid/tazobactam; ESBL: extended-spectrum beta-lactamases; Cephalosporins-ES: extended-spectrum cephalosporins; Cephalosporins-1G: first-generation cephalosporins.
The groups of special importance6,7 appear in boldface.
The ESBL are class A enzymes that degrade penicillins, cephalosporins (except cephamycins) and monobactams. They are often coded for by plasmid genes.8 These enzymes are commonly inhibited by clavulanic acid, tazobactam, sulbactam and new inhibitors7 (Fig. 1). From a structural point of view there are a very large number of families, with TEM, SHV (related to an intrinsic chromosomal enzyme — without ESBL profile — of Klebsiella pneumoniae) and CTX-M (in particular CTX-M-15 and CTX-M-14) being the most important.9,10 Among others, the clones of Escherichia coli with sequence type (ST) ST131 or of K. pneumoniae ST11 and ST405 are implicated in the worldwide spread of CTX-M-15.11–13
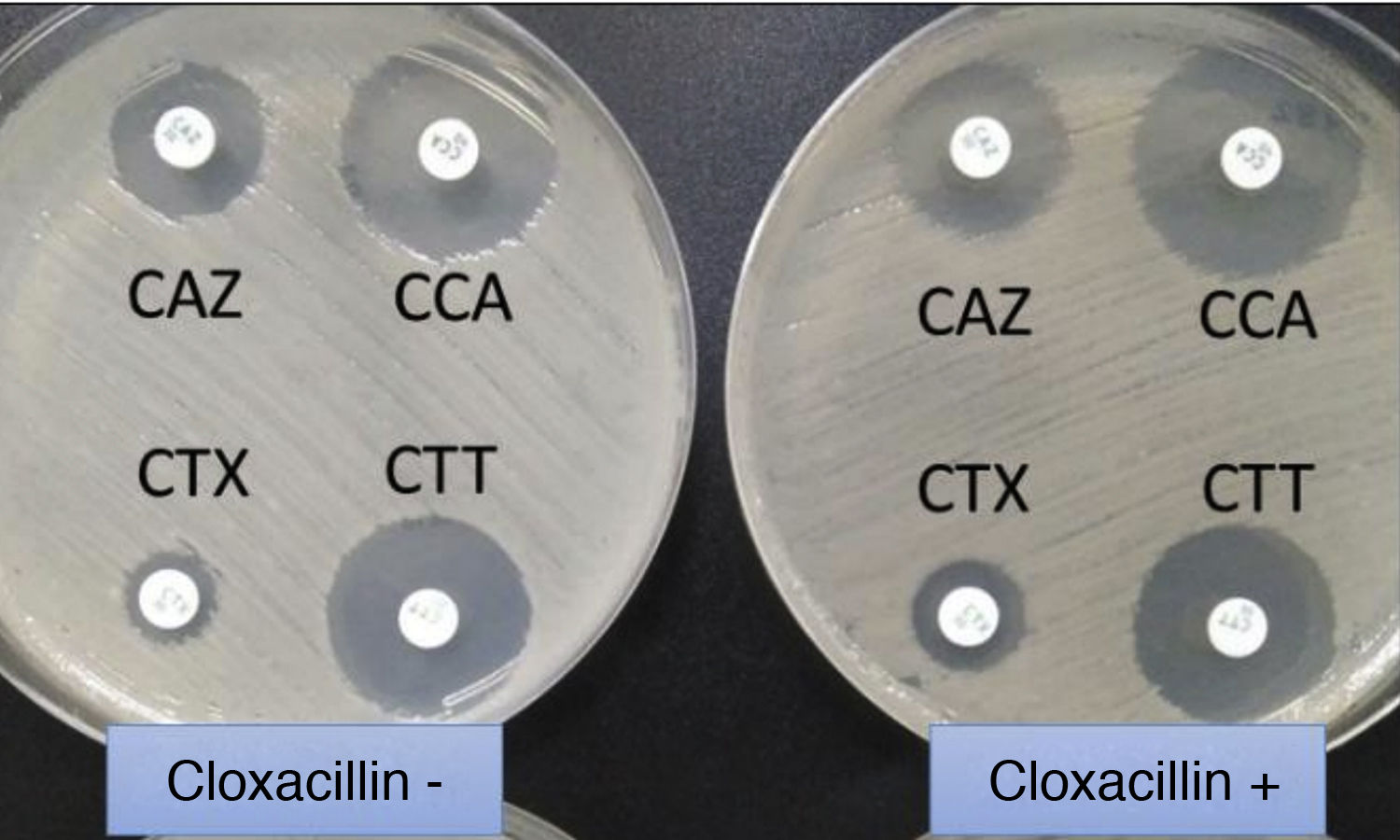
Extended-spectrum beta-lactamase (ESBL)-producing Klebsiella pneumoniae. In usual medium (without cloxacillin), the halos of ceftazidime (CAZ) and cefotaxime (CTX) are seen to be of smaller diameter than those of both cephalosporins combined with clavulanic acid (CCA and CTT, respectively). In medium with cloxacillin, which would inhibit the eventual additional presence of a class C beta-lactamase - plasmidic in the case of K. pneumoniae - no increase in the halos is observed, thus discarding this possibility.
The class C beta-lactamases include both AmpC enzymes coded for by chromosomal genes14 and variants coded for by plasmids (plasmidic cephamycinases).15 As a norm, they are not inhibited by clavulanic acid (neither tazobactam nor sulbactam), but are susceptible to new inhibitors such as avibactam.16 In many species (though not in E. coli), the production of chromosomal AmpC is regulated by a complex system of genes. Basal conditions are characterized by a low production of AmpC (repression state), though certain beta-lactams are more or less effective in inducing the production of the enzyme, and when the compound disappears, such induction ceases.16,17 Mutations in regulator genes17 can condition high-level production of enzyme even if there is no inducing beta-lactam; the corresponding strains are therefore referred to as derepressed strains.18 Most plasmidic cephamycinases are produced at a high level.15 The level of resistance of the microorganism to each beta-lactam depends on the amount of enzyme (beta-lactam induction capacity or enzyme derepression state) and the resistance of each compound to enzyme hydrolysis.14 The class C enzymes can degrade penicillins and cephalosporins such as cefotaxime and ceftazidime, and although they do not hydrolyze carbapenems effectively (with some exceptions, such as CMY-10 or ACT-28), when the enzyme is hyper-produced in strains with additional mechanisms (see below), clinically important resistance levels can be reached.19
The class A beta-lactamases with carbapenemase activity are mainly represented by the KPC family, and to a lesser extent by GES (not all their variants possess carbapenemase activity), SME, IMI and other variants.20–22 The KPC enzymes exhibit universal distribution, and generically hydrolyze carbapenems, penicillins, cephalosporins and monobactams. They are not inhibited by clavulanic acid (in fact, they hydrolyze the drug), but are inhibited by avibactam, vaborbactam and relebactam. To date, almost 100 variants of KPC are known, a number of which (e.g., KPC-31) produce resistance to ceftazidime-avibactam, though they do not efficiently hydrolyze carbapenems – thus generating a phenotype similar to that of the ESBLs23 (Fig. 2). The blaKPC gene forms part of transposon Tn4401 (of which several isoforms exist) and is vehiculized by different plasmids that also encode for genes of resistance to other families of antimicrobials. Several species of KPC-producing enterobacteria have been identified, though in view of their frequency, special mention must be made of the so-called high-risk clones of K. pneumoniae ST258, ST14, ST15, ST307, etc.24
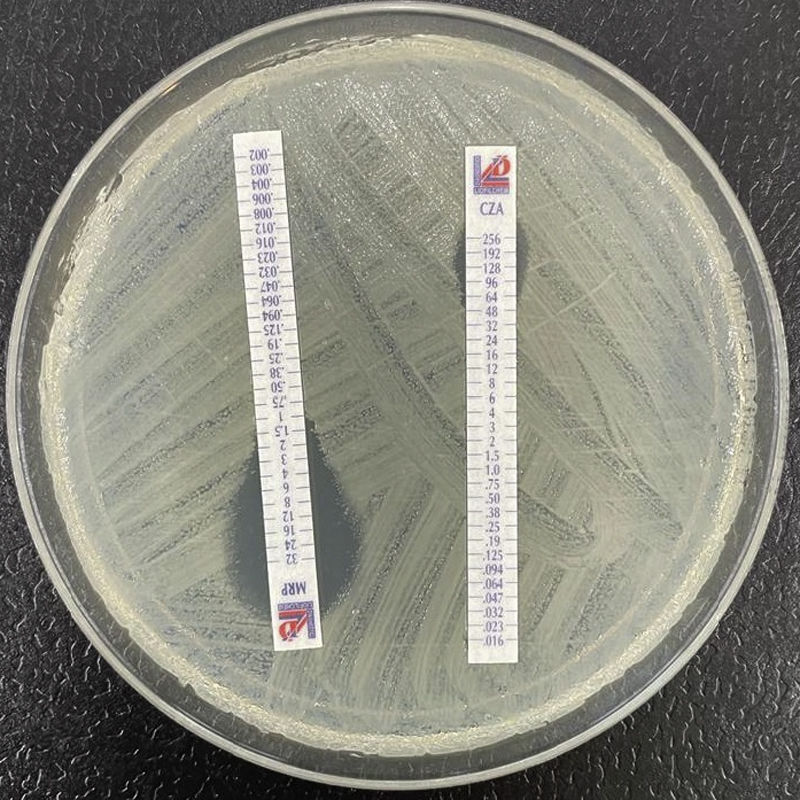
The class B beta-lactamases, or metallo-β-lactamases, degrade carbapenems and other compounds (penicillins and cephalosporins, but not monobactams) via a Zn2+ dependent mechanism. These enzymes are not inhibited by the currently available inhibitors. Although they are inhibited with EDTA (or other binding agents), this is of no practical use from the therapeutic perspective.25 Of the different families known, the NDM enzymes (with plasmidic or chromosomal coding) in enterobacteria are geographically widespread in isolates of different species (E. coli ST101 or ST131, K. pneumoniae, etc.). There have also been descriptions of strains that produce VIM, IMP or other families.
The class D enzymes are generically known as oxacillinases, because in vitro they hydrolyze this compound more efficiently than the class A or C enzymes.26,27 Structurally, they constitute a heterogeneous group of beta-lactamases that may behave as penicillinases, extended-spectrum beta-lactamases and carbapenemases. With some exceptions, they are not inhibited by clavulanic acid, tazobactam or sulbactam. Some of them (e.g., the OXA-48 group) are inhibited by avibactam. In Enterobacterales, the most relevant carbapenemase among the oxacillinases is OXA-48, initially identified in K. pneumoniae in Turkey, but with a widespread distribution in the Mediterranean setting and in other countries. In this context, blaOXA-48 is usually associated with a conjugative plasmid and with variants of Tn1999 found in different ST of K. pneumoniae and other species. Although OXA-48 has little capacity to hydrolyze extended-spectrum cephalosporins, many strains with this enzyme also usually produce CTX-M-15; they consequently exhibit resistance to these compounds.28 Other variants related to OXA-48 have been described, such as OXA-163 (with activity against extended-spectrum cephalosporins) or OXA-181.
In addition to the production of beta-lactamases, Enterobacterales resistance to beta-lactams is related to other mechanisms. Loss or structural alteration of the porins (hydrophilic channels through which the antimicrobials reach the interior of the bacteria) in itself causes a minor increase in resistance which – in the absence of other mechanisms – would have a limited clinical impact, but which on acting synergically with the latter would increase resistance. The impact is greater when one same strain loses its different porins: many ESBL or carbapenemase-producing clones of K. pneumoniae already lack porin OmpK35; as a result, the additional loss of the second main porin (OmpK36) results in a marked increase in resistance.29 The loss of porins is also relevant in other species, such as Enterobacter spp.19 Curiously, in E. coli, although multiple details are known about the role of porins in resistance in the laboratory strain K-12, less information is available when it comes to clinical isolates. The active efflux pumps, which eliminate beta-lactams once they have penetrated the bacteria, also contribute to increase resistance – though their role in the case of the beta-lactams of greatest clinical interest is less relevant than in other antimicrobials or in other microorganisms (as will be commented on addressing P. aeruginosa).30 The combination of porin loss, hyperproduction of the AcrB pump, and the hyper-expression of KPC-2 has been related to resistance to the new combination meropenem-vaborbactam.
In contrast to what is seen in gram-positive bacteria such as Staphylococcus aureus or Streptococcus pneumoniae, the role of alterations of the penicillin-binding proteins (PBPs) in Enterobacterales resistance to beta-lactams is small, though some studies have pointed to the interest of mutations in PBP-3.31
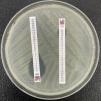
Resistance to quinolonesResistance to quinolones in Enterobacterales is related to multiple mechanisms. Traditionally, special attention has focused on resistance derived from alterations of the type II topoisomerases (topoisomerase II or DNA gyrase and topoisomerase IV). These occur as a consequence of mutations in the so-called quinolone resistance determining region (QRDR) of the gyrA (DNA gyrase) and parC genes (topoisomerase IV). Less important are the mutations in gyrB or parE. A single mutation in gyrA causes an increase in resistance to non-fluorinated quinolones (nalidixic acid) and a low level of resistance to fluorinated quinolones (Fig. 3), though the level of resistance to the latter rises parallel to the increase in the number of new mutations in gyrA and parC.32
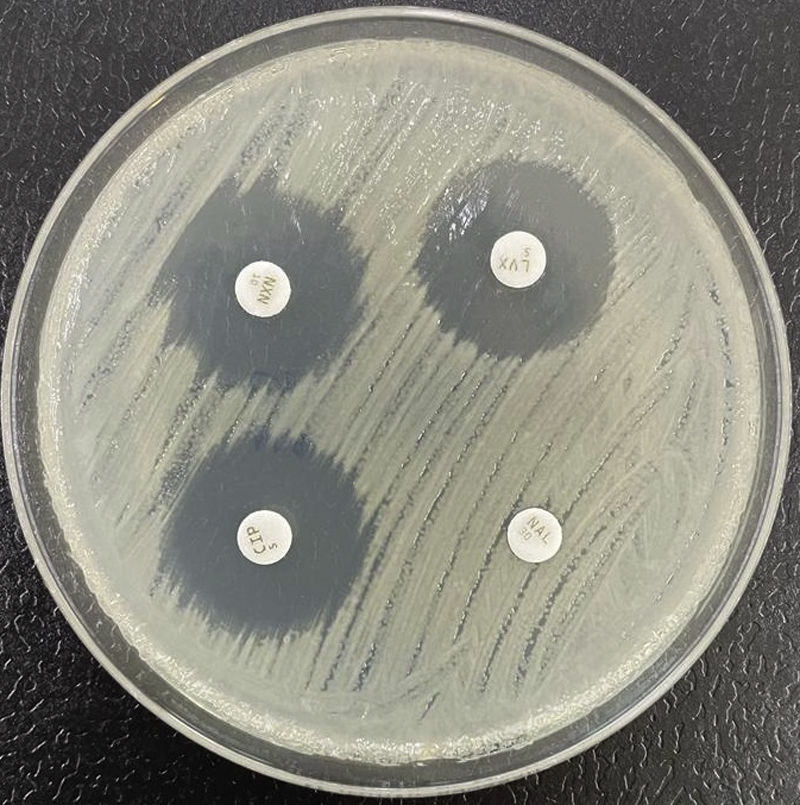
As in the case of beta-lactams, alterations of the porins contribute to increasing the level of resistance to quinolones. It has also been shown that lipopolysaccharide modifications are related to resistance to these compounds. With regard to the active efflux pumps, special interest focuses on those of the family RND (resistance-nodulation-division), which are structurally integrated into systems of three proteins: the pump itself, an efflux channel in the external membrane and a coupling protein of the other two. The RND systems eliminate a range of compounds, including quinolones. The best-studied example is that of AcrA-AcrB-TolC, the expression of which contributes to the (low) basal level of resistance30; this system can be hyper-expressed due to mutations in other regulator genes (operon mar, acrR, etc.), consequently raising the level of resistance. Some of these regulators (mar) are capable of simultaneously leading to loss of a porin and to hyperproduction of the efflux pump.33 The great majority of strains of K. pneumoniae possess the chromosomal oqxAB genes, which encode for an active efflux mechanism that expels hydrophilic quinolones and other compounds.
Several families of Qnr proteins34 coded for by plasmids have been identified that produce resistance to quinolones via a mechanism that protects the target of these compounds (topoisomerases). In different species, intrinsic qnr type chromosomal genes have even been identified. Following the discovery of Qnr, other plasmidic resistance mechanisms have been characterized, related to the production of acetylase AAC(6′)-Ib-cr, and affecting both (some) quinolones and (some) aminoglycosides, or to QepA (active efflux pump) expression. These plasmidic mechanisms per se produce (very) low levels of resistance, but when they accumulate within one same strain or coincide with chromosomal mechanisms, the levels exceed the clinical resistance cut-off points.35
Resistance to aminoglycosidesThe two fundamental mechanisms of resistance to aminoglycosides in Enterobacterales are the enzymatic modification of these compounds or the modification of their target.36,37 Additional information is also available on disorders in penetration into the bacteria or active elimination by efflux pumps (AcrD).30
The aminoglycoside-modifying enzymes are grouped into three large families of nucleotidyl (adenyl)-transferases, phosphotransferases or acetyltransferases, each with an infinity of variants, that respectively transfer AMP, phosphate or acetyl coenzyme A to certain positions of the aminoglycoside molecule, canceling the antibacterial effect of the drug.36
Each concrete enzyme affects certain aminoglycosides, but not others. In addition, one same microorganism can produce multiple enzymes of the same or of different families. As a result, the resistance phenotypes are difficult to correlate to single proteins (genes). To further increase complexity, there are two different nomenclature systems for these enzymes (for the proteins and for their genes). The enzymes most commonly found in Enterobacterales (most studies refer to multiresistant E. coli and K. pneumoniae) are AAC(3)-IIa, AAC(3)-IVa, AAC(6′)-Ib (with variants that affect the quinolones, as commented above), ANT(2″)-Ia, APH(3′)-Ia, APH(3′)-IIa, APH(3″)-Ib, ANT(2″)-Ia and ANT(3″)-Ia. In any case, it must be taken into account that certain enzymes affect compounds that are currently of little clinical interest (e.g., ANT3″-Ia only affects streptomycin and spectinomycin).36 The new compound plazomicin escapes practically all of the modifying enzymes of this kind.38
Ribosomal modification as a cause of resistance to aminoglycosides may be due to alterations of the proteins of the ribosome or – more importantly, due to their frequency – to the modification of specific sites in 16SrRNA mediated by methyl-transferases (methylases) coded for by plasmids.37 There are two large families of these methylases: N7-G1405 (ArmA and variants of Rmt) and N1-A1408 (NmpA). The two families inactivate the aminoglycosides of clinical interest (gentamycin, tobramycin, amikacin and even plazomicin), resulting in high-level resistance, and differ from each other in inactivating or not inactivating other compounds that are rarely used in current clinical practice. Much of the information available on this mechanism of resistance refers to the study of multiresistant strains; in Spain, it has recently been found that 5.1% of all carbapenemase-producing enterobacteria (mainly K. pneumoniae and E. cloacae) produce methylases, in particular RmtF.39
Resistance to polymyxinsA number of the enterobacteria most commonly isolated from clinical samples present intrinsic resistance to polymyxins, including Proteus spp., Morganella, Providencia spp., Serratia marcescens and Hafnia alvei.
Other species can develop resistance acquired through different mechanisms, of which the most important is the modification of lipopolysaccharide (LPS) due to the addition of different molecules – most commonly 4-amino-4-deoxy-l-arabinose or galactosamine (mediated by chromosomal genes) or phosphoethanolamine (mediated by chromosomal genes or by the recently discovered mcr type plasmid genes).40–42 Following this modification, the net negative charge of the LPS decreases, which complicates the interaction of the polymyxins with the bacteria. The comparative importance of other mechanisms, such as the production of capsule or active efflux pumps, is secondary.
The chromosomal genes that add the indicated compounds are activated as a consequence of mutations in double-component systems (composed of a sensor transmembrane histidine-kinase protein that undergoes auto-phosphorylation under certain conditions, and another cytoplasmic protein that on undergoing phosphorylation modulates the expression of different genes). These systems include PhoP-PhoQ, PmrA-PmrB and CrrA-CrrB.40–42 In K. pneumoniae, mutations of MgrB (a small transmembrane protein that under normal conditions negatively regulates the kinase activity of the PhoP/PhoQ system) are one of the main causes of resistance to polymyxins.43
The family of mcr plasmid genes codes for phosphoethanolamine transferases that modify LPS in a way analogous to other chromosomal genes. Their expression does not always result in levels that exceed the clinical resistance cut-off points.41
Non-fermenting gramnegative bacilliPseudomonas aeruginosaResistance to betalactamsPseudomonas aeruginosa possesses a type AmpC chromosomal cephalosporinase (Fig. 4), and its constitutive expression characterizes the natural resistance of P. aeruginosa to aminopenicillins (including the combination amoxicillin-clavulanate), first- and second-generation cephalosporins, some third-generation cephalosporins (cefotaxime, ceftriaxone) and ertapenem.44 The ureidopenicillins (piperacillin, ticarcillin), some third-generation cephalosporins (ceftazidime) and the fourth-generation cephalosporin, cefepime, are not affected by the basal expression of this beta-lactamase.
Penicillins and cephalosporins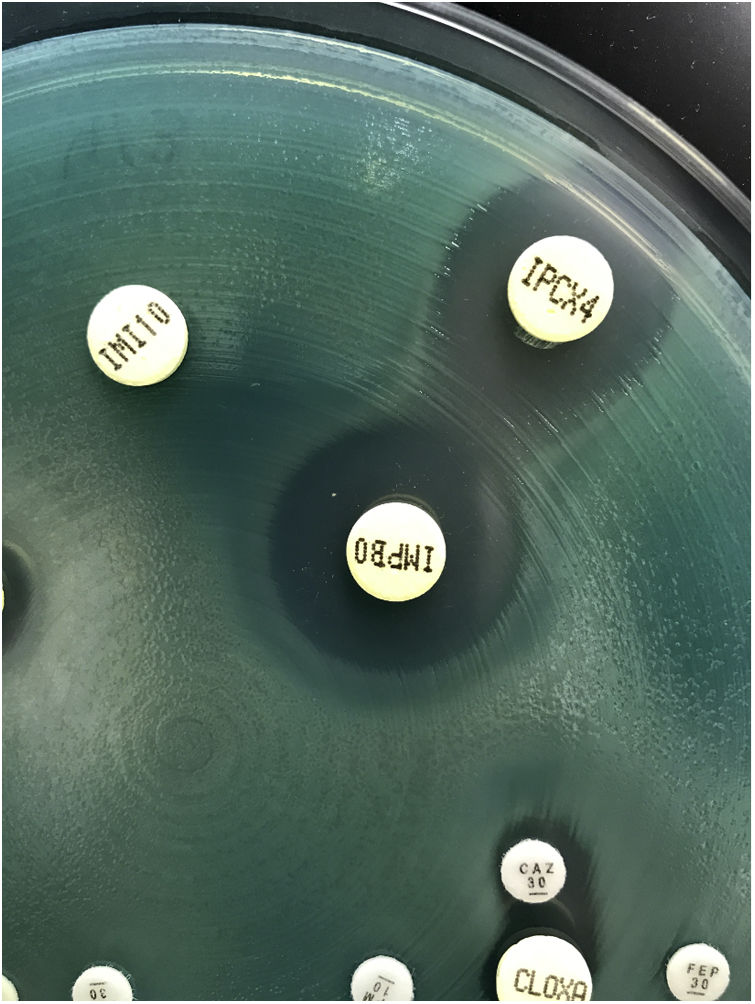
Pseudomonas aeruginosa has a strong capacity to develop resistance through mutation, fundamentally the selection of mutants with constitutive hyperproduction of its chromosomal cephalosporinase AmpC45 – this being the main mechanism of resistance to the penicillins (piperacillin, piperacillin-tazobactam) and cephalosporins (ceftazidime and cefepime). Such hyperproduction, together with structural modifications of the chromosomal cephalosporinase AmpC, is the principal mechanism of clinical resistance to ceftolozane/tazobactam and ceftazidime/avibactam in vivo.46,47
On the other hand, the hyper-expression of any of its multiple efflux pumps, mainly MexAB-OprM, MexXY-OprM and MexCD-OprJ, contributes significantly to the cephalosporin resistance phenotypes. The mutational hyper-expression of MexAB-OprM affects cefepime and ceftazidime equally, and that of MexXY-OprM and MexCD-OprJ affects cefepime.48
Pseudomonas aeruginosa can also exhibit resistance to cephalosporins mediated by extended-spectrum beta-lactamases (ESBL) vehiculized in plasmids and/or integrons that affect sensitivity to ceftazidime and cefepime, though not uniformly so. The type GES, PER, TEM, SHV and VEB beta-lactamases mainly possess ceftazidimase activity, affecting cefepime to a lesser extent, and some GES type ESBLs can affect the carbapenems.49 The OXA type ESBLs can exhibit ceftazidimase or cefepimase activity, depending on the type,50 and the development of OXA-10 resistance to ceftolozane/tazobactam and ceftazidime/avibactam has recently been described.51
CarbapenemsResistance to carbapenems in P. aeruginosa is generally associated with chromosomal mutations that alter its porins, efflux pump overexpression, intrinsic beta-lactamase derepression, or a combination of these.52 However, these mechanisms do not affect imipenem and meropenem uniformly.
The main mechanism of resistance to imipenem is the repression or inactivation of the porin OprD, which together with the inducible expression of its cephalosporinase AmpC, increases its basal MIC to values of 8−32 mg/l53 (Fig. 3). Nevertheless, meropenem not only uses OprD, but also employs other alternative entry pathways – this resulting in a more discrete increase in MIC, with values of 2−4 mg/l.54 Hyper-expression of the MexAB-OprM pump plus the inactivation of OprD is the most common cause of clinical resistance to meropenem.55
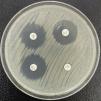
To a lesser extent, resistance to carbapenems can be mediated by acquired carbapenemases, mainly metallo-β-lactamases, which can hydrolyze most of the beta-lactam antibiotics. The genes that code for these metallo-β-lactamases are generally found in integrons that often carry additional genes which code for resistance to non-beta-lactam antibiotics. The most frequent and widespread metallo-β-lactamases are types VIM and IMP, and to a lesser degree NDM56 (Fig. 5). As mentioned above, some type GES ESBLs exhibit carbapenemase activity.
Resistance to fluoroquinolones
High-level resistance to fluoroquinolones is due to the interaction of mutations in the topoisomerases, including DNA gyrase (GyrA and GyrB) and topoisomerase IV (ParC and ParE), associated with hyper-expression through mutation of the efflux pumps MexAB/XY/CD/EF.49
Low-level resistance to quinolones is generally mediated by plasmids and is usually associated with AAC(6′)-Ib-cr modifying enzymes57.
Resistance to aminoglycosidesResistance to aminoglycosides is generally mediated by three mechanisms: enzymatic modification, active expulsion (efflux) mechanisms and the methylation of RNA16s.
The enzymatic modification mechanisms are frequently coded for by plasmids and can involve three types of enzymes: acetyltransferases (AAC), adenyltransferases (ANT) and phosphoryltransferases (APH). These enzymes do not affect all aminoglycosides uniformly. The most prevalent include AAC(69)-II, AAC(3)-II and ANT(29)-I, which determine resistance to gentamycin and tobramycin, while AAC(3)-I is associated with resistance to gentamycin.58
Other mechanisms include reduction of the intracellular concentration of aminoglycosides due to changes in the permeability of the external membrane or efflux pumps (MexAB-OprM),59 and to RNA16s methylation mechanisms mediated by methylases coded in transposons inserted in plasmids, which confer high-level resistance to all the aminoglycosides.60
In addition, there have been descriptions of gradual aminoglycoside MIC increments due to non-enzymatic mechanisms associated with mutation of the LPS or external membrane proteins.
Resistance to polymyxinsThe development of resistance to colistin generally implies modification of the LPS mediated by mutations in the double-component systems code for by pmrAB, phoPQ or parRS genes.61
Similarly, the inducible expression of the operon arnBCADTEF, responsible for the addition of a residual 4-aminoarabinose group to lipid A of the LPS, is fundamental for the development of inducible and/or adaptive resistance to colistin.62
Acinetobacter baumanniiResistance to betalactams- •
Cephalosporins. Acinetobacter baumannii exhibits low sensitivity to cephalosporins, since it produces a non-inducible type AmpC cephalosporinase and type OXA-51 oxacillinase; this, together with a low-level constitutive expression of one or more of its efflux pumps,63 confers intrinsic resistance to first- and second-generation cephalosporins, and to some third-generation cephalosporins such as cefotaxime and ceftriaxone.64 Resistance to the rest of the broad-spectrum cephalosporins may be mediated by the overexpression of these intrinsic resistance mechanisms. The overexpression of its intrinsic AmpC cephalosporinase due to an ISAba-1 insertion sequence leads to high-level resistance to ceftazidime and cefepima.65 The mutation-induced overexpression of the AdeABC efflux pump also confers high-level resistance to cephalosporins.66
On the other hand, resistance to cephalosporins can be mediated by the acquisition of plasmidic beta-lactamases, fundamentally types PER, VEB and GES, but also ESBLs of types TEM and SHV.67 This mechanism produces high-level resistance to ceftazidime and cefepime.
- •
Carbapenems. Resistance to carbapenems in A. baumannii is related to numerous beta-lactamases with carbapenemase activity, including type OXA carbapenemases – both constitutive, such as OXA-51/69 (hyper-produced by insertion of the sequence ISAba1), or acquired, fundamentally oxacillinases of the OXA-23 group, the OXA-24/40, OXA-58 and OXA-143 groups, type IMP, VIM, SIM and NDM metallo-β-lactamases or type GES ESBLs.68 These mechanisms lead to high-level resistance to imipenem and meropenem, with MIC values in the range of 16−32 mg/l. In some cases, resistance is associated with reduced expression of its external membrane proteins.
Furthermore, overexpression of the adeB gene, regulated by the adeRS genes (double-component regulator system of the AdeABC family of efflux pumps), also contributes to resistance to carbapenems, increasing the MIC values of imipenem and meropenem approximately two-fold.69
Resistance to quinolonesResistance to fluoroquinolones is generally the result of chromosomal mutations affecting quinolone resistance-determining regions of DNA gyrase (GyrA and GyrB) and topoisomerase IV (ParC and ParE) – sequentially resulting in high-level resistance to ciprofloxacin and levofloxacin.70 Furthermore, resistance can also be mediated by efflux pumps such as AdeABC and AdeM,71 which result in lesser level MIC increments. In this case, not only the fluoroquinolones but also the aminoglycosides and tetracyclines are affected.
Resistance to aminoglycosidesResistance can be mediated by several mechanisms: a) efflux pumps; b) target alterations; and 3) aminoglycoside modifying enzymes.72 Among the efflux pumps, the principal representative is AdeABC (also implicated in resistance to quinolones), which affects sensitivity to gentamycin, tobramycin and amikacin. A second pump, AbeM, mainly affects gentamycin.
The production of RNA16 s methylases is mediated by plasmids; the main representative is ArmA, which confers high-level resistance to gentamycin, tobramycin and amikacin (MIC > 256 mg/l). With regard to the aminoglycoside modifying enzymes, mention must be made of plasmidic acetyltransferases, nucleotidyl transferases and/or phosphotransferases, either alone or in combination, fundamentally the AAC(6́) family, which in their different variants affect gentamycin, tobramycin and/or amikacin (leading to high-level resistance)(MIC ≥ 32 mg/l).
Resistance to tetracyclinesResistance to first-generation tetracyclines (tetracycline), second-generation tetracyclines (doxycycline, minocycline) and tigecycline (structural analogue of minocycline) is mediated by efflux pumps.
Resistance to first- and second-generation tetracyclines is generally associated with efflux pumps coded for by the tetA and tetB genes located in transposons. The tetA gene is responsible for resistance to tetracycline and doxycycline, but not to minocycline, while the tetB gene has been found in isolates that were also resistant to minocycline.73
Resistance to tigecycline is mainly associated with hyper-expression of the AdeABC efflux pump. Another efflux pump (AdeIJK) could act synergically with AdeABC.74
Resistance to polymyxinsTwo mechanisms of resistance to colistin have been reported in A. baumannii: a) alterations in lipid A of the LPS as the result of mutations in the PmrAB double-component system75; and b) complete loss of LPS production resulting from mutations in the lpxA, lpxC and lpxD genes that code for the enzymes that catalyze the first steps in LPS biosynthesis.76 Resistance to colistin mediated by the plasmidic transmission mrc gene has not been described in A. baumannii.77
Stenotrophomonas maltophiliaStenotrophomonas maltophilia intrinsically presents a multiresistant phenotype related to the low permeability of its external membrane, which contains few porins,78 and to the presence of an SmeDEF efflux pump - affecting beta-lactams, quinolones and aminoglycosides.79 In addition, it is naturally resistant to aminoglycosides due to the presence of a chromosomal acetyltransferase, AAC(6′)-Iz.80 It also produces two inducible chromosomal beta-lactamases: carbapenemase L1, which confers intrinsic resistance to all the carbapenems, and cephalosporinase L2, which moreover also hydrolyzes aztreonam.78
The fluoroquinolones, and in particular levofloxacin, are active despite the low-level expression of a Qnr protein coded for at chromosomal level. High-level resistance to the fluoroquinolones may result from the selection of mutants with greater expression of the smQnr gene or of the SmeDEF efflux pump.81
Although S. maltophilia is very sensitive to the combination trimethoprim - sulfamethoxazole, resistance to the combination may appear through the acquisition of sul and dfrA genes, vehiculized by plasmids and class 1 integrons that code for dihydropteroate synthases with activity upon the sulfonamides.82
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Lepe JA, Martínez-Martínez L. Mecanismos de resistencia en bacterias gramnegativas. Med Intensiva. 2022;46:392–402.

